Трисомии и причины их возникновения
Одной из наиболее актуальных проблем современной медицинской генетики является определение этиологии и патогенеза наследственных заболеваний. Цитогенетические и молекулярные исследования имеют высокую диагностическую информативность и ценность при решении данной проблемы, так как хромосомные аномалии встречаются с частотой от 4 до 34% при различных наследственных синдромах.
Хромосомные синдромы – большая группа патологических состояний, возникающих в результате аномалии количества и/или структуры хромосом человека. Клинические проявления при хромосомных нарушениях наблюдаются с рождения и не имеют прогредиентного течения, поэтому правильнее называть эти состояния синдромами, а не заболеваниями.
Частота хромосомных синдромов составляет 5-7 на 1000 новорожденных. Аномалии хромосом достаточно часто возникают, как в половых, так и в соматических клетках человека.
В работе рассматриваются наследственные синдромы вызванные численными мутациями хромосом – трисомии (трисомия 21 – синдром Дауна, трисомия 18 – синдром Эдвардса, трисомия 13 – синдром Патау, трисомия 8 – синдром Варкани, трисомия X 947, XXX).
Целью работы является: изучение цитогенетических и клинических проявлений трисомий, возможных рисков и методов диагностики.
причина проявление трисомия человек
ГЛАВА 1 ЧИСЛЕННЫЕ ХРОМОСОМНЫЕ МУТАЦИИ
Анеуплоидия (др.-греч. ἀν- — отрицательная приставка + εὖ — полностью + πλόος — попытка + εἶδος — вид) — наследственное изменение, при котором число хромосом в клетках не кратно основному набору. Может выражаться, например, в наличии добавочной хромосомы (n + 1, 2n + 1 и т. п.) или в нехватке какой-либо хромосомы (n — 1, 2n — 1 и т. п.). Анеуплоидия может возникнуть, если в анафазе I мейоза гомологичные хромосомы одной или нескольких пар не разойдутся.
В этом случае оба члена пары направляются к одному и тому же полюсу клетки, и тогда мейоз приводит к образованию гамет, содержащих на одну или несколько хромосом больше или меньше, чем в норме. Это явление известно под названием нерасхождение.
Когда гамета с недостающей или лишней хромосомой сливается с нормальной гаплоидной гаметой, образуется зигота с нечетным числом хромосом: вместо каких-либо двух гомологов в такой зиготе их может быть три или только один. (1, 10)
Зигота, в которой количество аутосом меньше нормального диплоидного, обычно не развивается, но зиготы с лишними хромосомами иногда способны к развитию. Однако из таких зигот в большинстве случаев развиваются особи с резко выраженными аномалиям.
Формы анеуплоидии:
Моносомия — это наличие всего одной из пары гомологичных хромосом. Примером моносомии у человека является синдром Тернера, выражающийся в наличии всего одной половой (X) хромосомы. Генотип такого человека X0, пол — женский. У таких женщин отсутствуют обычные вторичные половые признаки, характерен низкий рост и сближенные соски. Встречаемость среди населения Западной Европы составляет 0,03 %. (2, 10)
В случае обширной делеции в какой-либо хромосоме иногда говорят о частичной моносомии, например синдром кошачьего крика.
Трисомия — Трисомией называют появление в кариотипе дополнительной хромосомы. Самым известным примером трисомии является болезнь Дауна, которую часто называют трисомией по хромосоме 21. Результатом трисомии по хромосоме 13 является синдром Патау, а по хромосоме 18 — синдром Эдвардса. Все названные трисомии — аутосомные. Другие трисомики по аутосомам нежизнеспособны, погибают внутриутробно и, по-видимому, теряются в виде спонтанных абортов. Жизнеспособными являются индивидуумы с дополнительными половыми хромосомами. Более того, клинические проявления дополнительных хромосом X или Y могут быть весьма незначительными. (2)
Другие случаи нерасхождения аутосом:
Трисомия 16 выкидыш
Трисомия 9 Трисомия 8 (синдром Варкани). (3)
Случаи нерасхождения половых хромосом:
XXX (женщины без фенотипических особенностей, у 75% наблюдается умственная отсталость различной степени, алалия. Нередко недостаточное развитие фолликулов в яичниках, преждевременное бесплодие и ранний климакс (необходимо наблюдение эндокринолога). Носительницы ХХХ плодовиты, хотя риск спонтанных абортов и хромосомных нарушений у потомства у них несколько повышен по сравнению со средними показателями; частота проявления 1:700) (1, 2, 3)
XXY, Синдром Клайнфельтера (мужчины, обладающие некоторыми вторичными женскими половыми признаками; бесплодны; яички развиты слабо, волос на лице мало, иногда развиваются молочные железы; обычно низкий уровень умственного развития)
XYY: мужчины высокого роста с различным уровнем умственного развития. (3)
Тетрасомия и пентасомия
Тетрасомия (4 гомологичные хромосомы вместо пары в диплоидном наборе) и пентасомия (5 вместо 2-х) встречаются чрезвычайно редко. Примерами тетрасомии и пентасомии у человека могут служить кариотипы XXXX, XXYY, XXXY, XYYY, XXXXX, XXXXY, XXXYY, XYYYY и XXYYY. Как правило, с нарастанием количества "лишних" хромосом увеличивается тяжесть и выраженность клинических симптомов. (5,7)
Характер и тяжесть клинических симптомов при различных типах хромосомных перестроек, определяются степенью нарушения генетического баланса и, как следствие, гомеостаза в организме человека. Можно отметить лишь некоторые общие закономерности клинических проявлений хромосомных синдромов.
Недостаток хромосомного материала приводит к более выраженным клиническим проявлениям, чем его избыток. Частичные моносомии (делеции) по определенным участкам хромосом сопровождаются более тяжелыми клиническими проявлениями, чем частичные трисомии (дупликации), что обусловлено потерей ряда генов, необходимых для роста и дифференцировки клеток. В этом случае структурные и количественные перестройки хромосом, в которых локализованы гены, экспрессирующиеся в раннем эмбриогенезе, часто оказываются летальными и обнаруживаются у абортусов и мертворожденных. К гибели эмбриона на ранней стадии развития приводят полные моносомии по аутосомам, а также трисомии по 1, 5, 6, 11 и 19 хромосомам. Наиболее часто встречаются трисомии по хромосомам 8, 13, 18 и 21. (3, 6, 7)
Для большинства хромосомных синдромов, обусловленных аномалиями аугосом, характерны пренатальная гипотрофия (малый вес ребенка при доношенной беременности), пороки развития двух и более органов и систем, а также задержка темпов раннего психомоторного развития, олигофрения и снижение показателей физического развития ребенка. У детей с хромосомной патологией часто выявляют увеличение количества, так называемых, стигм дизэмбриогенеза или малых аномалий развития. В случае наличия пяти и более таких стигм говорят о повышении порога стигматизации у человека. К стигмам дизэмбриогенеза можно отнести наличие сандалевидной щели между первым и вторым пальцами на ногах, диастему (увеличение расстояния между передними резцами), расщепление кончика носа и другие. (2, 4, 5)
Для аномалий половых хромосом, в противоположность аугосомным синдромам, не характерно наличие выраженного интеллектуального дефицита, некоторые больные имеют нормальное или даже выше среднего умственное развитие. У большинства больных с аномалиями половых хромосом возникает бесплодие и невынашивание беременности. Необходимо отметить, что бесплодие и самопроизвольное прерывание беременности при аномалиях половых хромосом и аугосом имеет различные причины. При аномалиях аутосом прерывание беременности часто обусловлено наличием хромосомных перестроек, несовместимых с нормальным эмбриональным развитием, или элиминацией несбалансированных по хромосомному материалу зигот, эмбрионов и плодов. При аномалиях половых хромосом в большинстве случаев наступление беременности и ее вынашивание невозможно по причине аномалии сперматозоидов или аплазии или резкой гипоплазии, как наружных, так и внутренних половых органов. В целом, аномалии половых хромосом приводят к возникновению менее выраженных клинических симптомов, чем аномалии аутосом. (7)
Тяжесть клинических проявлений зависит от соотношения нормального и аномального клеточных клонов. (6, 7)
Полные формы хромосомных аномалий характеризуются более тяжелыми клиническими проявлениями, чем мозаичные.
Таким образом, учитывая все клинико-генетические и генеалогические данные больных с хромосомными синдромами, показания к исследованию кариотипа у детей и взрослых следующие:
• малый вес новорожденного при доношенной беременности;
• врожденные пороки развития двух и более органов и систем;
• врожденные пороки развития двух и более органов и систем в сочетании с олигофренией;
• недифференцированная олигофрения;
• бесплодие и привычное невынашивание беременности;
• наличие сбалансированной хромосомной перестройки у родителей или сибсов пробандов. (3,4)
ГЛАВА 2.КЛИНИКО-ГЕНЕТИЧЕСКИЕ ХАРАКТЕРИСТИКИ ТРИСОМИЙ
Наиболее распространенный тип количественных аномалий хромосом -трисомии и тетрасомии по одной из пар. У живорожденных чаще всего встречаются трисомии по 8, 9, 13, 18, 21 и 22 аутосомам. При возникновении трисомии по другим аугосомам (особенно большим метацентрическим и субметацентрическим), эмбрион оказывается нежизнеспособным и гибнет на ранних сроках внутриутробного развития. Летальный эффект имеют и моносомии по всем аугосомам. (2, 3)
Выделяют два онтогенетических варианта трисомий: транслокационный и регулярный. Первый вариант достаточно редко выступает в качестве этиологического фактора и составляет не более 5% всех случаев трисомий по аутосомам. Транслокационные варианты синдромов хромосомных трисомий могут появляться у потомков носителей сбалансированных хромосомных перестроек (чаще всего, робертсоновских или реципрокных транслокаций и инверсий), а также возникать de novo. (3)
Остальные 95% случаев трисомий по аутосомам представлены регулярными трисомиями. Существует две основные формы регулярных трисомий: полная и мозаичная. В подавляющем большинстве случаев (до 98%) обнаруживаются полные формы, возникновение которых может быть обусловлено, как гаметическими мутациями (нерасхождением или анафазным отставанием хромосомы при мейотическом делении одной единственной гаметы), так и наличием сбалансированных хромосомных перестроек во всех клетках родителей.
В редких случаях наследование количественных хромосомных перестроек происходит от родителей, имеющих полную форму трисомии (например, по Х- или 21-хромосоме).
Мозаичные формы трисомий составляют около 2% всех случаев и характеризуются различным соотношением нормальных и трисомных клеточных клонов, которое и определяет вариабельность клинических проявлений.
Приводим основные клинико-цитогенетические характеристики трех наиболее распространенных вариантов полных трисомий по аутосомам у человека. (3)
Обычно трисомии возникают из-за нарушения расхождения гомологичных хромосом в анафазе мейоза I. В результате в одну дочернюю клетку попадают обе гомологичные хромосомы, а во вторую дочернюю клетку не попадает ни одна из хромосом бивалента (такую клетку называют нулисомной). Иногда, однако, трисомия может быть результатом нарушения расхождения сестринских хроматид в мейозе II. В этом случае в одну гамету попадают две совершенно одинаковые хромосомы, что в случае ее оплодотворения нормальным спермием даст трисомную зиготу. Этот тип хромосомных мутаций, ведущих к трисомии, называют нерасхождением хромосом. Отличия в исходах нарушения расхождения хромосом в мейозе I и II иллюстрирует рис. 1. Аутосомные трисомии возникают из-за нерасхождения хромосом, наблюдающегося преимущественно в оогенезе, но и в сперматогенезе нерасхождение аутосом также может быть. Нерасхождение хромосом может происходить и на ранних стадиях дробления оплодотворенной яйцеклетки. В этом случае в организме присутствует клон мутантных клеток, который может захватывать большую или меньшую часть органов и тканей и иногда давать клинические проявления, сходные с теми, которые наблюдают при обычной трисомии.(1, 4)
Причины нерасхождения хромосом остаются неясными. Известный факт связи между нерасхождением хромосом (особенно хромосомы 21) и возрастом матери до сих пор не имеет однозначной интерпретации. Некоторые исследователи полагают, что это может быть связано со значительным промежутком времени между конъюгацией хромосом и образованием хиазм, которые происходят у плода женского пола, т.е. достаточно рано и с расхождением хромосом в диакинезе, наблюдающемся у женщин в детородном возрасте. Следствием старения ооцитов могут быть нарушение образования веретена и другие нарушения механизмов завершения мейоза I. Рассматривается также версия об отсутствии образования хиазм в мейозе I у плодов женского пола, которые необходимы для последующего нормального расхождения хромосом. (2)
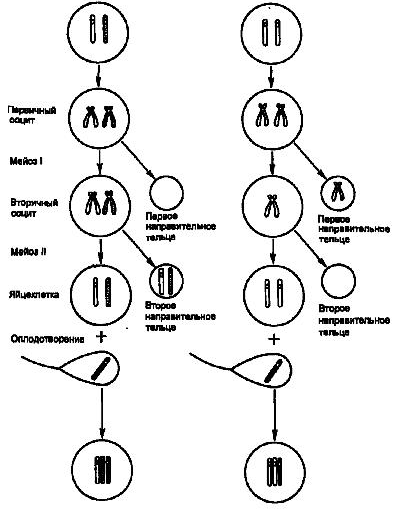
Нерасхождение в мейозе I Нерасхождение в мейозе II
Рис. 1. Мейотическое нерасхождение
ГЛАВА 3. ТРИСОМИЯ ПО 21-Й ХРОМОСОМЕ, ИЛИ СИНДРОМ ДАУНА
3.1 Цитогенетическая характеристика синдрома Дауна
Самой частой из трисомий и вообще одной из самых частых наследственных болезней являетсятрисомия 21, или синдром Дауна. Цитогенетическая природа синдрома Дауна была установлена Ж. Леженом в 1959 г. Синдром встречается в среднем с частотой 1 на 700 живорожденных, но частота синдрома зависит от возраста матерей и повышается с его увеличением. У женщин старше 45 лет частота рождения больных с синдромом Дауна достигает 4 %. (2)
Цитогенетическими причинами синдрома Дауна являются регулярная трисомия — 95 %, транслокации хромосомы 21 на другие хромосомы — 3 % и мозаицизм — 2 %. Молекулярно- генетические исследования позволили выявить критический район хромосомы 21, ответственный за основные клинические проявления синдрома Дауна, — 21q22. (2, 4, 7)
Причиной синдрома Дауна также может быть робертсоновская транслокация. Если вовлечены хромосомы 21 и 14, что случается нередко, то в результате может образоваться зигота с трисомией по хромосоме 21, которая приведет к рождению ребенка с болезнью Дауна. Для робертсоновских транслокаций с участием хромосомы 21 риск рождения такого ребенка составляет 13 %, если носителем транслокации является мать, и 3 %, если носитель — отец. Возможность рождения ребенка с болезнью Дауна у родителей с робертсоновской транслокацией, в которой участвует хромосома 2/, надо постоянно иметь в виду, так как риск повторного рождения больного ребенка разный при регулярной трисомии 21, обусловленной нерасхождением хромосом, и трисомии 21, связанной с носитель- ством робертсоновской транслокации одним из родителей. В том случае, когда робертсоновская транслокация является результатом слияния длинных плеч хромосом 21, все гаметы будут несбалансированными: 50 % будет иметь две хромосомы 21 и 50 % будет нулисомной по хромосоме 21. В семье, в которой один из родителей является носителем такой транслокации, все дети будут с болезнью Дауна. (16)
Повторный риск при регулярной трисомии 21составляет примерно 1:100 и зависит от возраста матери. При семейной транслокации показатели риска варьируют от 1 до 3 %, если носителем транслокации является отец, и от 10 до 15 %, если носителем транслокации является мать. Как уже отмечалось, при редких случаях транслокации 21q21q повторный риск составляет 100 %. (2)
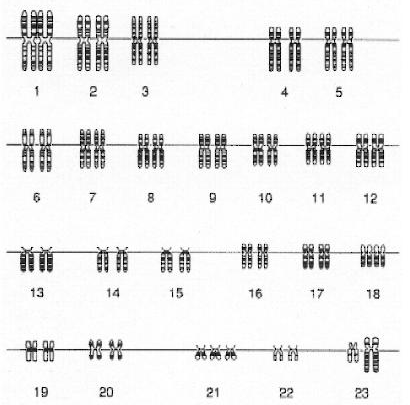
Рис. 2 Схематическое изображение кариотипа мужчины, страдающего синдромом Дауна. Нерасхождение хромосом G21 в одной из гамет привело к трисомии по этой хромосоме
Таким образом, Цитогенетические варианты синдрома Дауна разнообразны. Однако основную долю (94—95%) составляют случаи простой полной трисомии 21 как следствие нерасхождения хромосом в мейозе. При этом материнский вклад нерасхождения в эти гаметические формы болезни составляет 80%, а отцовский — только 20%. Причины такой разницы неясны Небольшая (около 2%) доля детей с синдромом Дауна имеет мозаичные формы (47+21/46). Примерно 3—4% больных с синдромом Дауна имеют транслокационную форму грисомии по типу робертсоновских транслокаций между акроиентриками (D/21 и G/21). Почти 50% транслокационных форм наследуется от родителей-носителей и 50% — транслокации, возникшие de novo. (1)
Соотношение мальчиков и девочек среди новорождённых с синдромом Дауна составляет 1:1. (1)
3.2 Клинические проявления синдрома Дауна
Синдром Дауна, трисомия 21, — наиболее изученная хромосомная болезнь. Частота синдрома Дауна среди новорождённых равна 1:700—1:800, не имеет какой-либо временной, этнической или географической разницы у родителей одинакового возраста. Частота рождения детей с синдромом Дауна зависит от возраста матери и в меньшей мере от возраста отца (рис. 3). (8)
С возрастом существенно возрастает вероятность рождения детей с синдромом Дауна. Так, в возрасте 45 лет она составляет около 3%. Высокая частота детей с синдромом Дауна (около 2%) наблюдается у рано рожающих женщин (до 18 лет). Следовательно, для популяционных сравнении частоты рождения детей с синдромом Дауна надо принимать во внимание распределение рожающих женщин по возрасту (доля женщин, рожающих после 30—35 лет, среди всех рожающих). Это распределение меняется иногда в течение 2—3 лет для одного и того же населения (например, при резком изменении экономической ситуации в стране). В связи с уменьшением в 2 раза числа женщин, рожающих после 35 лет, в последние 15 лет в Белоруссии и России число детей с синдромом Дауна снизилось на 17—20%. Увеличение частоты с увеличением материнского возраста известно, но в то же время необходимо понимать, что большинство детей с синдромом Дауна рождены матерями, возраст которых младше 30 лет. Это связано с большим числом беременностей в этой возрастной группе по сравнению со старшей группой.
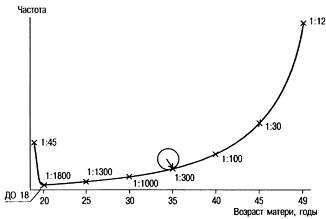
Рис. 3 Зависимость частоты рождения детей с синдромом Дауна от возраста матери
В литературе описана «пучковость» рождения детей с синдромом Дауна в определённые промежутки времени в некоторых странах (городах, провинциях).
Эти случаи можно объяснить скорее стохастическими колебаниями спонтанного уровня нерасхождения хромосом, чем воздействием предполагаемых этиологических факторов (вирусная инфекция, низкие дозы рааиации, хлорофос). (1)
Клиническая симптоматика синдрома Дауна разнообразна: это и врождённые пороки развития, и нарушения постнатального развития нервной системы, и вторичный иммунодефицит и др.
Дети с синдромом Дауна рождаются в срок, но с умеренно выраженной пренатальной гипоплазией (на 8—10% ниже средних величин). Многие симптомы синдрома Дауна заметны при рождении, в последующем они проявляются более чётко. Квалифицированный педиатр ставит правильный диагноз синдрома Дауна в родильном доме не менее чем в

Рис. 4 Дети разного возраста с характерными чертами синдрома Дауна (брахицефалия, круглое лицо макроглоссия и открытый рот эпикант, гипертелоризм, широкая переносица, косоглазие)
90% случаев. Из черепно-лицевых дизморфий отмечаются монголоидный разрез глаз (по этой причине синдром Дауна долго называли монголоидизмом), круглое уплощённое лицо, плоская спинка носа, эпикант, крупный (обычно высунутый) язык, брахицефалия, деформированные ушные раковины (рис. 4).

На трех рисунках представлены фотографии детей разного возраста, и у всех имеются характерные черты и признаки дизэмбриогенеза.
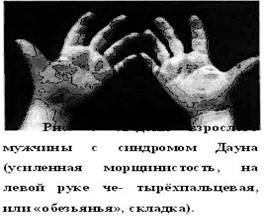
Характерна мышечная гипотония в сочетании с разболтанностью суставов (рис. 5). Часто встречаются врождённый порок сердца, клинодактилия, характерные изменения дерматоглифики (четырёхпальцевая, или «обезьянья», складка на ладони — рис. 5.6, две кожные складки вместо трёх на мизинце, высокое положение трирадиуса и др.). Пороки ЖКТ наблюдаются редко. Частота какого-либо симптома в 100% случаев, кроме низкого роста, не отмечена. В табл. 5.2 и 5.3 представлена частота внешних признаков синтрома Дауна и основных врождённых пороков внутренних органов. (1)
Диагноз синдрома Дауна ставится на основании частоты сочетания нескольких симптомов ( табл. 1 и 2). Следующие 10 признаков наиболее важны для постановки диагноза, наличие 4—5 из которых достоверно указывает на синдром Дауна: 1) уплощение профиля лица (90%); 2) отсутствие сосательного рефлекса (85%); 3) мышечная гипотония (80%); 4) монголоидный разрез глаз (80%); 5) избыток кожи на шее (80%); 6) разболтанность суставов (80%); 7) диспластичный таз (70%); 8) диспластичные (деформированные) ушные раковины (40%); 9) клинодактилия мизинца (60%); 10) четырёхпальцевая сгибательная складка (поперечная линия) на ладони (40%). Большое значение для диагностики имеет динамика физического и умственного развития ребёнка. При синдроме Дауна и то и другое задерживается. Рост взрослых больных на 20 см ниже среднего. Задержка в умственном развитии достигает имбецильнос- ти, если не применяются специальные методы обучения. Дети с синдромом Дауна ласковые, внимательные, послушные, терпеливые при обучении. Коэффициент умственного развития (10) у разных детей широко варьирует (от 25до75).(1,2,3) Реакция детей с синдромом Дауна на факторы окружающей среды часто патологическая в связи со слабым клеточным и гуморальным иммунитетом, снижением репарации ДНК, недостаточной выработкой пищеварительных ферментов, ограниченными компенсаторными возможностями всех систем. По этой причине дети с синдромом Дауна часто болеют пневмониями, тяжело переносят детские инфекции. У них отмечается недостаток массы тела, выражен авитаминоз.
Таблица 1. Наиболее частые внешние признаки синдрома Дауна (по Г. И. Лазюку с доп.)
| Порок и.ш признак | Частота, % общего числа больных |
| Мозговой череп и лицо | 98,3 |
| Брахицефалия | 81,1 |
| Монголоидный разрез глазных щелей | 79,8 |
| Эпикант | 51,4 |
| Плоская спинка носа | 65,9 |
| Узкое нёбо | 58,8 |
| Большой высунутый язык | 9 |
| Деформированные ушные раковины | 43,2 |
| Костно-мышечная. система, конечности | 100,0 |
| Низкий рост | 100,0 |
| Деформация грудной клетки | 26,9 |
| Короткие и широкие кисти | 64,4 |
| Клинодактилия мизинца | 56,3 |
| Укороченная средняя фаланга V пальца кисти с одной сгибательной складкой | ? |
| Четырёхпальцевая складка на ладони | 40,0 |
| Сандалевидная щель | ? |
| Глаза | 72,1 |
| Пятна Брашфилда | 68,4 |
| Помутнение хрусталика | 32,2 |
| Косоглазие | 9 |
Таблица2. Основные врождённые пороки внутренних органов при синдроме Дауна (по Г. И. Лазюку с дополнениями)
| Пораженная система и порок | Частота % общего числа больных |
| Сердечно-сосудистая система | 53,2 |
| Дефект межжелудочковой перегородки | 31,4 |
| Дефект межпредсердной перегородки | 24,3 |
| Открытый атриовентрикулярный канал | 9 |
| Аномалии крупных сосудов | 23,1 |
| Органы пищеварения | 15,3 |
| Атрезия или стеноз двенадцатиперстной кишки | 6,6 |
| Атрезия пищевода | 0,9 |
| Атрезия прямой кишки и ануса | 1,1 |
| Мегаколон | 1,1 |
| Мочевая система (гипоплазия почек, гидроуретер, гидронефроз) | 5,9 |
Врождённые пороки внутренних органов, сниженная приспособленность детей с синдромом Дауна часто приводят к летальному исходу в первые 5 лет.
Следствием изменённою иммунитета и недостаточности репарационных систем (для повреждённой ДНК) являются лейкозы, часто встречающиеся у больных с синдромом Дауна. (8, 11)
Дифференциальная диагностика проводится с врождённым гипотиреозом, другими формами хромосомных аномалий. Цитогенетическое исследование у детей показано и при подозрении на синдром Дауна, и при клинически установленном диагнозе, поскольку цитогенетическая характеристика пациента необходима для прогноза здоровья будущих детей у родителей и их родственников. (14)
Этические проблемы при синдроме Дауна многоплановы. Несмотря на повышение риска рождения ребёнка с синдромом Дауна и другими хромосомными синдромами, врач должен избегать прямых рекомендаций по планированию беременности у женщин старшей возрастной группы, так как возрастной риск остаётся достаточно низким, особенно с учётом возможностей пре- натальной диагностики.
Неудовлетворённость у пациентов часто вызывает форма сообщения о синдроме Дауна у ребёнка. Поставить диагноз синдрома Дауна по фенотипическим признакам обычно можно немедленно после родо- разрешения. Врач, пытающийся отказаться от установления диагноза до исследования кариотипа, может потерять уважение родственников ребёнка. Важно сообщить родителям по крайней мере о ваших подозрениях как можно скорее после родоразрешения. Нецелесообразно полностью информировать родителей ребёнка с синдромом Дауна немедленно после родоразрешения. Нужно дать достаточно сведений, чтобы ответить на их немедленные вопросы и поддерживать их до того дня, когда станет возможно более детальное обсуждение. Немедленная информация должна включать объяснение этиологии синдрома для исключения взаимных обвинений супругов и описание исследований и процедур, необходимых для того, чтобы полностью оценить здоровье ребёнка. (1, 14)
Полное обсуждение диагноза нужно провести, как только родители, по крайней мере частично, оправятся от стресса родоразрешения, обычно в пределах 1-х суток. К этому времени у них возникает комплекс вопросов, на которые необходимо отвечать точно и определённо. На эту встречу приглашают обоих родителей. В этот период ещё слишком рано нагружать родителей всей информацией о заболевании, так как эти новые и сложные понятия требуют времени для восприятия. (1,8)
Не пытайтесь делать прогнозы. Бесполезно пробовать с точностью предвидеть будущее любого ребёнка. Древние мифы типа «по крайней мере он будет всегда любить и наслаждаться музыкой» непростительны. Важно отметить, что способности каждого ребёнка развиваются индивидуально. (1, 2, 3)
Лечебная помощь детям с синдромом Дауна многопланова и неспецифична. Врождённые пороки сердца устраняют оперативно. Постоянно проводится общеукрепляющее лечение. Питание должно быть полноценным. Необходимы внимательный уход за больным ребёнком, защита от действия вредных факторов окружающей среды (простуда, инфекции). Многие больные с три- сомией 21 теперь способны вести самостоятельную жизнь, овладевают несложными профессиями, создают семьи. (1)
ГЛАВА 3. СИНДРОМ ЭДВАРДСА – ТРИСОМИЯ 18
При цитогенетическом исследовании обычно обнаруживают регулярную трисомию 18. Как и при синдроме Дауна, выявляется связь между частотой трисомии 18и возрастом матери. В большинстве случаев дополнительная хромосома имеет материнское происхождение. Около 10 % трисомии 18 обусловлены мозаицизмом или несбалансированными перестройками, чаще робертсоновскими транслокациями. (2)

Рис. 7 Кариотип Трисомия 18
Клинических различий между цитогенетически различающимися формами трисомии нет. (1)
Частота синдрома Эдвардса составляет 1:5000—1:7000 новорождённых. Соотношение мальчиков и девочек равно 1:3. Причины преобладания больных девочек пока неясны.
При синдроме Эдвардса отмечается выраженная задержка пренатального развития при полной продолжительности беременности (роды в срок). На рис. 8-9 представлены пороки развития, характерные для синдрома Эдвардса. В первую очередь это множественные врождённые пороки развития лицевой части черепа, сердца, костной системы, половых органов. (1,2)
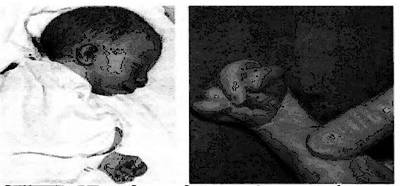
Рис. 8 Новорожденный с Рис. 9 Характерное для синдромом Эдвардса. синдрома Эдвардса Выступающий затылок; положение пальцев микрогения; флексорное (возраст ребенка 2 мес.) положение кисти
Череп долихоцефалической формы; нижняя челюсть и отверстие рта маленькие; глазные щели узкие и короткие; ушные раковины деформированные и низко расположенные. Из других внешних признаков отмечаются флексорное положение кистей, аномально развитая стопа (пятка выступает, сводно провисает), I палец стоп короче II. Спинномозговая грыжа и расщелина губы встречаются редко (5% случаев синдрома Эдвардса).
Многообразная симптоматика синдрома Эдвардса у каждого больного проявляется лишь частично. Частота отдельных врожденных пороков приведена в табл. 3. (1)
Таблица3. Основные врождённые пороки при синдроме Эдвардса (по Г. И. Лазюку)
| Пораженная система и порок (признак) | Относительная частота, % |
| Мозговой череп и лицо | 100,0 |
| Ультразвуки в живой природе Учения о составе вещества. Процесс фотосинтеза. Влияние солнечной активности на биосферу Ферменты дереворазрушающих грибов Физиология центральной нервной системы
Актуально:
|