Физико-химические основы адсорбционной очистки воды от органических веществ
Объем потребляемой в мире воды достигает 4 трлн. м3 в год, а
преобразованию со стороны человека подвергается практически вся гидросфера. Химическая и нефтехимическая отрасли промышленности способствуют проникновению в водную среду веществ, обычно отсутствующих в ней, или превышению естественного уровня их концентрации, ухудшающей качество водной среды. (9)
За время существования человечества в природную среду было введено огромное количество органических веществ. Вредные химические элементы и вещества попадают в водоемы, ухудшая их санитарное состояние и вызывая необходимость специальной глубокой очистки воды перед использованием ее для хозяйственно-питьевых и некоторых промышленных целей. Многие органические примеси не извлекаются из воды механически, не нейтрализуются при биологической очистке, не удаляются такими традиционными методами водоочистки, как отстаивание, коагуляция и флотация. Это обуславливает введение в комплексную технологическую схему водоподготовки стадии адсорбционной доочистки. Как правило, эта стадия является заключительным этапом в технологическом процессе очистки воды. Адсорбционный метод является хорошо управляемым процессом. Он позволяет удалять загрязнения различного характера практически до любой остаточной концентрации независимо от их химической устойчивости. При этом отсутствуют вторичные загрязнения. Отсюда перспективной является тенденция развития фильтрующе-сорбирующих устройств, предназначенных для локальной доочистки питьевой воды, и этот вопрос является весьма актуальным.
2. АДСОРБЦИОННЫЕ МЕТОДЫ
Адсорбция – процесс концентрирования растворенного вещества (адсорбата) или растворителя (адсорбтива) из объема фаз на поверхности раздела между ними (на поверхности твердого тела (адсорбента) или жидкости). (3)
В зависимости от характера сорбционного взаимодействия адсорбата и адсорбента различают физическую адсорбцию, активированную адсорбцию и хемосорбцию.
Физическая адсорбция обусловлена силами межмолекулярного взаимодействия Ван-дер-Ваальса, не избирательна, полностью необратима, протекает с высокой скоростью и имеет сравнительно низкую теплоту адсорбции – от нескольких килоджоулей до нескольких десятков килоджоулей на моль адсорбата. Адсорбция протекает молекулярно, т.е. преимущественно адсорбируются соединения в недиссоциированном состоянии. Физическая адсорбция характерна для веществ, адсорбируемых из парогазовой фазы, а при адсорбции из растворов осложнена физико-химическим взаимодействием адсорбата, адсорбтива и адсорбента.
Активированная адсорбция обусловлена взаимодействием адсорбата и адсорбента с образованием поверхностного соединения особого рода, характерного тем, что молекулы адсорбента, вступившие во взаимодействие с молекулами адсорбата (адсорбтива), остаются в кристаллической решетке адсорбента. Активированная адсорбция избирательна, как правило, протекает медленно (с повышение температуры скорость адсорбции заметно возрастает), необратима и характеризуется высокой теплотой адсорбции – до нескольких сотен килоджоулей на моль адсорбата.
Хемосорбция – обычная химическая реакция, протекающая на поверхности адсорбента и сопровождающаяся выделением теплоты, эквивалентной теплоте химической реакции.
Использование комбинации нескольких адсорбентов разного типа позволяет осуществлять комплексную корректировку состава воды по всем необходимым в каждом конкретном случае показателям. Использование смесей адсорбентов определенного состава для подготовки питьевой воды не только высокоэффективно, но и чрезвычайно экономически выгодно. (4)
3. Сорбция из водных растворов
Материал, на поверхности пор которого происходит концентрирование поглощаемого вещества, называют адсорбентом, а само вещество — адсорбатом. Адсорбционные явления основаны на физическом и химическом взаимодействии адсорбата и адсорбента.
Силы молекулярного взаимодействия, в основном, дисперсионные, обуславливающие физическую адсорбцию, возникают при сближении молекул материала адсорбента и адсорбируемого вещества и проявляются в упорядочении движения частиц вследствие взаимного притяжения. Дисперсионные взаимодействия неспецифичны, присущи всем веществам и различаются в конкретных случаях лишь количественно. Потенциальная энергия взаимодействия двух атомов равна
![]() (1)
(1)
где r— расстояние между центрами атомов; — эмпирическая константа; Сn — константа поляризации.
Приведенное выражение показывает, что адсорбционные взаимодействия проявляются только на очень малых расстояниях. Теоретически, если взаимное притяжение атомов максимально на расстоянии r0, то при ![]() их взаимодействие ослабевает в 4 — 5 раз. И, наоборот, при
их взаимодействие ослабевает в 4 — 5 раз. И, наоборот, при ![]() превалируют силы отталкивания. Иногда дисперсионные взаимодействия усиливаются водородными связями и электростатическими (индукционными или ориентационными) силами. Эти дополнительные взаимодействия специфичны для определенного вида адсорбируемых веществ или свойств поверхности адсорбента. Адсорбция – процесс самопроизвольный, протекающий с выделением некоторого количества теплоты,
превалируют силы отталкивания. Иногда дисперсионные взаимодействия усиливаются водородными связями и электростатическими (индукционными или ориентационными) силами. Эти дополнительные взаимодействия специфичны для определенного вида адсорбируемых веществ или свойств поверхности адсорбента. Адсорбция – процесс самопроизвольный, протекающий с выделением некоторого количества теплоты,
В отличие от физической адсорбции, носящей обратимый характер с сохранением индивидуальности адсорбата и адсорбента, хемосорбция — скорее химический процесс на границе раздела фаз. Этот процесс обычно необратимый и значительно более экзотермичный, чем физическая сорбция.
Сорбция из жидких растворов значительно сложнее, чем из парогазовой смеси, так как включает взаимодействия сорбента с сорбируемым веществом и с растворителем (водой); при этом также следует учитывать взаимодействие растворителя с сорбатом. Поэтому, несмотря на то, что сорбция из водных растворов исследуется и используется почти 200 лет, она изучена значительно менее сорбции из парогазовой фазы. В основном механизм сорбции из растворов в том или ином виде объясняют представлениями, выведенными для газовой фазы, лишь дополняя или ограничивая условиями, специфическими для жидкой фазы. Отличия в подходе к подобному переносу отражаются на виде и точности моделей и расчетов систем сорбционной очистки воды.
Теория полимолекулярной сорбции, в основе которой лежит представление о многослойной сорбции на поверхности макро- и мезопористого сорбента, была разработана Брунауэром, Эметом и Теллером. Однако в практике наибольшее распространение получили микропористые сорбенты, в чрезвычайно малом пространстве микропор которых послойной сорбции вещества на поверхности не происходит. При сорбции в микропорах происходит заполнение части или всего объема их сорбатом, который под действием взаимно усиливающихся и перекрывающихся адсорбционных полей, создаваемых противоположными стенками пор, находится в специфическом уплотненном состоянии. Теории объемного заполнения микропор, разработанная Дубининым и его школой, использует понятие о предельном объеме адсорбционного пространства микропористого сорбента W0. Основное уравнение адсорбции паров и газов на микропористых сорбентах, известное как уравнение Дубинина – Радушкевича, имеет вид:
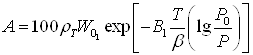 (2)
(2)
При сорбции вещества из раствора оно занимает на поверхности или в объеме пор сорбента место, которое до этого занимали молекулы растворителя (воды), а не свободное пространство. Присутствие воды в порах приводит к некоторому выравниванию сорбционного потенциала. В объеме сорбируемой фазы концентрация вещества значительно выше, чем в растворе. При этом снижается поверхностное натяжение на границе раздела раствор – твердый сорбент.
Основы термодинамики адсорбции из растворов впервые сформулировал Гиббс более 100 лет назад. Им было введено понятия избыточной адсорбции ![]() , т.е. избыточного содержания поглощаемого вещества в адсорбированной фазе по сравнению с его содержанием в растворе. Величина избыточной (гиббсовской) адсорбции легко определима по формуле
, т.е. избыточного содержания поглощаемого вещества в адсорбированной фазе по сравнению с его содержанием в растворе. Величина избыточной (гиббсовской) адсорбции легко определима по формуле
![]() (3)
(3)
где С0 и Ск – концентрация вещества в растворе до и после опыта; V – объем раствора; m – навеска сорбента.
Термодинамический подход к решению проблемы сорбции является наиболее общим и позволяет оценивать сорбируемость молекул по значению максимальной работы переноса вещества из раствора на поверхность сорбента. Поскольку при сорбции вещества из воды происходит уменьшение свободной энергии системы ![]() , Когановский предложил использовать эту величину для прогнозирования эффективности извлечения растворенных соединений из воды. Константа равновесия при сорбции из разбавленных растворов
, Когановский предложил использовать эту величину для прогнозирования эффективности извлечения растворенных соединений из воды. Константа равновесия при сорбции из разбавленных растворов ![]() связана с
связана с ![]() зависимостью вида
зависимостью вида ![]() , из которой следует, что чем больше
, из которой следует, что чем больше ![]() , тем лучше сорбируется вещество.
, тем лучше сорбируется вещество.
Идею о возможности применения теории объемного заполнения микропор для описания сорбции из жидкой фазы выдвинули Эльтеков и Стадник. Эта идея использует представления об отсутствии влияния физического состояния сорбата в объемной фазе на сорбционный потенциал в микропорах углей и отсутствии ассоциативных, ионных и водородных связей между молекулами сорбируемого вещества и воды, а также внутри сорбата. Эта теория применима для расчета сорбции из очень разбавленных растворов ограничено растворимых веществ: уравнение изотермы сорбции на микропористых АУ в данном случае принимает вид:
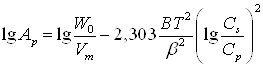 (4)
(4)
Здесь W0 – предельный объем микропор сорбента; Vm – мольный объем сорбата; ![]() – структурно-энергетическая константа;
– структурно-энергетическая константа; ![]() – растворимость соединения в воде при данной температуре;
– растворимость соединения в воде при данной температуре; ![]() – равновесная концентрация в воде.
– равновесная концентрация в воде.
Обобщая данные о сорбируемости на АУ низкомолекулярных органических соединений, можно сделать вывод, что менее других сорбируются структурно простые вещества в ионной форме, лучше – в молекулярной. Сорбируемость органических веществ возрастает в ряду:
гликоли < спирты < кетоны < сложные эфиры < альдегиды <
< недиссоциированные кислоты < ароматические соединения.
Вода, сорбируясь на участках окисленной поверхности АУ, препятствует сорбции на них неполярных алифатических соединений. Сорбируемость органических веществ возрастает с увеличением длины углеродной цепи (если она не ограничена размером пор сорбента), причем для гомологов, согласно правилу Траубе, на величину, кратную изменению длины углеродной цепи.
Способность к сорбции возрастает с ростом молекулярной массы органических веществ, особенно выше 30 000. Аналогична и зависимость при росте мицеллярной массы коллоидов. Присутствие в воде неорганических солей, способствующих укрупнению ассоциатов молекул красителей и гуматов, приводит к их более полному сорбционному выделению.
Специфическими загрязнениями сточных вод заводов СК являются неокисляемые биохимическими путем лейканол и трилон Б. Первый обладает свойствами ПАВ и присутствует в воде в виде отдельных молекул и ассоциатов, что и определяет особенность сорбции его из воды, а именно: при 20–30ºС отдельные молекулы и ассоциаты малых размеров сорбируются на ГАУ, а крупные ассоциаты способны лишь к дальнейшему укрупнению и коагуляции. Поэтому сорбционная емкость ГАУ зависит от начальной концентрации лейканола в воде, так как пропорционально росту ![]() увеличивается и доля крупных ассоциатов лейканола.
увеличивается и доля крупных ассоциатов лейканола.
Эффективность сорбции трилона Б на БАУ, ОУ, АГ-3, СКТ при ![]() ммоль/дм3 почти одинакова. Сорбция трилона Б из воды – типичный случай полимолекулярного поглощения с преобладающим взаимодействием во втором и последующих слоях над взаимодействием в первом слое.
ммоль/дм3 почти одинакова. Сорбция трилона Б из воды – типичный случай полимолекулярного поглощения с преобладающим взаимодействием во втором и последующих слоях над взаимодействием в первом слое.
Применение угля СКТ более эффективно в области малых равновесных концентраций (![]() ммоль/дм3); при больших
ммоль/дм3); при больших ![]() целесообразнее сорбция на АГ-3.
целесообразнее сорбция на АГ-3.
Такие биологические объекты, как бактерии и вирусы, несмотря на крупные размеры, хорошо сорбируются АУ. Изотерма сорбции E. Coli описывается уравнением Лэнгмюра, а вируса Polio – уравнением Фрейндлиха.
Влияние температуры на сорбцию из водных растворов далеко не однозначно. Дело в том, что при сорбции на микропористых сорбентах веществ, размеры молекул которых близки к эффективным размерам пор, проникновение этих молекул в поры зависит от их кинетической энергии. При достаточной энергии (температуре) молекулы сорбата проникают в окна пор и сорбируются; в противном случае происходит лишь незначительно поглощение на поверхности мезо- и макропор. Иными словами, сорбционная емкость повышается с ростом температуры; это явление получило название «активированной адсорбции». В то же время физическая сорбция, как любой экзотермический процесс, в целом ухудшается с ростом температуры. Поэтому суммарное внешне фиксируемое проявление этих двух явлений (активированной и физической адсорбции) может иметь экстремум при определенной температуре. Как показано в работах Дубинина, для газов и паров этот экстремум имеет вид острого пика.
При сорбции из водных растворов, а в особенности из таких многокомпонентных систем, как сточные воды, этот пик размыт. Например, при доочистке сточных вод максимальные значения сорбционной емкости достигались при 35 – 60ºС. Емкость угля при 0 – 10 и выше 75 – 80ºС была в 1,5 – 2 раза ниже. В других случаях оптимальная температура может быть иной. Есть данные о том, что с повышением температуры значительно улучшается кинетика адсорбции при доочистке высококонцентрированных вод, что связано с перестройкой молекул в растворах, разрушением ассоциатов воды и сорбируемого вещества, снижением вязкости раствора. Изменение сорбируемости различных веществ на АУ при нагревании неодинаково.
Многокомпонентность состава природных и сточных вод приводит к тому, что показатели сорбции малого (до 5%) или очень большого (более 95%) количества загрязнений от общего отличаются от средних. Обычно это выражается в завышенных показателях сорбции небольшой части хорошо сорбируемых примесей и, наоборот, в низких показателях при высокой степени очистки от плохо сорбируемых веществ. Последнее приводит к тому, что при сорбции в статических условиях при необходимости повысить эффект очистки воды до 92 – 95% дозу АУ следует резко увеличить. (5)
4. Изотермы адсорбции. Уравнение Лэнгмюра. Уравнение фрейндлиха
Основные сведения о сорбционных свойствах материала и характере адсорбции на нем определенных веществ могут быть получены из изотерм адсорбции, характеризующих зависимость сорбционной способности ![]() от концентрации
от концентрации ![]() (или давления
(или давления ![]() ) сорбируемого компонента при постоянной температуре:
) сорбируемого компонента при постоянной температуре: ![]() для жидкой фазы или
для жидкой фазы или ![]() для газов. Брунауэр, Эммет и Теллер выделили пять основных типов изотерм сорбции (рис 1). Выпуклые участки изотерм /, II и IV типов (Лэнгмюровская адсорбция) указывают на наличие в сорбентах микропор, но, кроме того, сорбенты II и IV имеют еще и макропоры. Изотермы III и V типов встречаются реже и описывают сильное межмолекулярное взаимодействие в веществе сорбата. Крутизна изотермы Iтипа характеризует размер микропор сорбентов: а — ультрамикропористых, б — микропористых. Изотерма !Vб принадлежит переходно-пористому сорбенту; 1Vв — однородно макропористому, а 1Vа — со смешанной структурой.
для газов. Брунауэр, Эммет и Теллер выделили пять основных типов изотерм сорбции (рис 1). Выпуклые участки изотерм /, II и IV типов (Лэнгмюровская адсорбция) указывают на наличие в сорбентах микропор, но, кроме того, сорбенты II и IV имеют еще и макропоры. Изотермы III и V типов встречаются реже и описывают сильное межмолекулярное взаимодействие в веществе сорбата. Крутизна изотермы Iтипа характеризует размер микропор сорбентов: а — ультрамикропористых, б — микропористых. Изотерма !Vб принадлежит переходно-пористому сорбенту; 1Vв — однородно макропористому, а 1Vа — со смешанной структурой.
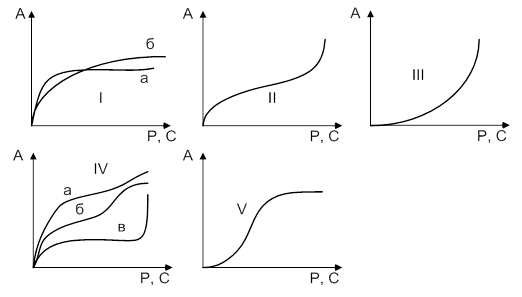
Рис. 1. Типы изотерм сорбции по БЭТ
Чем круче изотерма, тем мельче микропоры. Предельное значение такой адсорбции Апр соответствует покрытию поверхности мономолекулярным слоем. Вогнутые участки указывают на наличие макропор. (4)
Изотерма мономолекулярной адсорбции на микропористых сорбентах обычно имеет вид, показанный на рис. 2. Эта кривая имеет два прямолинейных участка – при малых и больших значениях равновесной концентрации адсорбата. Как адсорбция паров, так и адсорбция из растворов осуществляется в области пор с радиусом 0,5 – 1,6 нм.
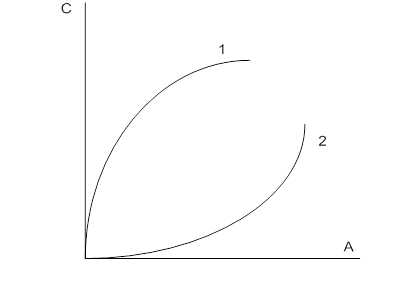
Рис.2. Изотермы адсорбции:
1 – выпуклая (Лэнгмюра);
2 – вогнутая (на макропорах)
Для аналитического описания изотермы мономолекулярной адсорбции чаще всего используется уравнение Лэнгмюра:
![]() (5)
(5)
где ![]() – удельная адсорбция, ммоль/г:
– удельная адсорбция, ммоль/г: ![]() – предельная адсорбция, ммоль/г;
– предельная адсорбция, ммоль/г; ![]() – равновесная концентрация адсорбата, ммоль/л;
– равновесная концентрация адсорбата, ммоль/л; ![]() – адсорбционная константа.
– адсорбционная константа.
Уравнение Лэнгмюра описывает изотерму адсорбции во всех областях равновесных концентраций. При малых концентрациях, когда ![]() , формула упрощается и приобретает вид:
, формула упрощается и приобретает вид:
![]() (6)
(6)
Для условий, когда ![]() , формула (5) дает независимость адсорбции от концентрации:
, формула (5) дает независимость адсорбции от концентрации:
![]() (7)
(7)
Уравнение (5) может быть преобразовано в линейную форму путем его умножения на ![]() и последующего деления на
и последующего деления на ![]() , в результате чего оно приобретает вид:
, в результате чего оно приобретает вид:
 (8)
(8)
Уравнение (8) представляет собой уравнение прямой. По этой зависимости определяют величину предельной адсорбции (рис.3).
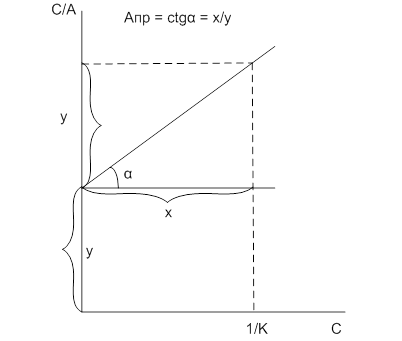
Рис. 3. Схема определения констант в уравнении Лэнгмюра
В области промежуточных равновесных концентраций (на небольших участках изменения концентрации адсорбата) зависимость адсорбции от концентрации часто может быть описана уравнением Фрейндлиха, в основе которого лежит допущение, что изотерма адсорбции является параболой:
![]() (9)
(9)
где ![]() и
и ![]() – константы. Константа
– константы. Константа ![]() зависит от природы адсорбента и адсорбата и колеблется в широких пределах. Константа
зависит от природы адсорбента и адсорбата и колеблется в широких пределах. Константа ![]() возрастает с увеличением длины углеводородного радикала ПАВ, т.к. при этом возрастает адсорбционная способность вещества. Показатель
возрастает с увеличением длины углеводородного радикала ПАВ, т.к. при этом возрастает адсорбционная способность вещества. Показатель ![]() колеблется в пределах 0,1 – 1 и зависит от температуры и природы адсорбата.
колеблется в пределах 0,1 – 1 и зависит от температуры и природы адсорбата.
В логарифмированном виде уравнение Фрейндлиха представляется прямой (рис. 13):
![]() (10)
(10)
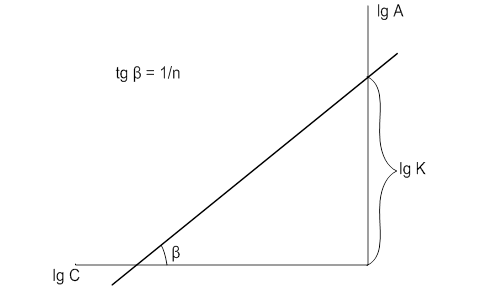
Рис. 4. Схема определения констант в уравнении Фрейндлиха
Отрезок, отсекаемый прямой на оси ординат, равен ![]() , а тангенс угла наклона прямой к оси абсцисс равен
, а тангенс угла наклона прямой к оси абсцисс равен ![]() . (3)
. (3)
5. Расчет изотермы адсорбции молекулярно-растворенных органических веществ на активных углях без экспериментальных измерений
Одна из основных задач при определении технологического режима адсорбционной установки – это расчет условий адсорбционного равновесия, т.е. расчет изотермы адсорбции. Зависимость количества адсорбированного вещества от равновесной концентрации в растворе необходимо знать при расчете кинетики и динамики адсорбции, а также при расчете удельного расхода адсорбента при любом способе технологического оформления адсорбционного процесса.
Большой интерес представляет расчет изотермы адсорбции без экспериментальных измерений. В основу такого расчета положены следующие представления:
– адсорбция органических веществ (неэлектролитов и слабых электролитов) на углеродных адсорбентах, являющаяся физическим нелокализованным процессом, обусловлена дисперсионным взаимодействием молекул органических веществ с углеродными атомами поверхности, и поэтому поверхностные оксиды не влияют на адсорбционное равновесие и избирательность адсорбции;
– общий объем всех адсорбированных компонентов раствора является величиной постоянной, не зависящей от структуры молекул, соотношения концентраций и их ориентации. Молярные объемы адсорбированных веществ приблизительно равны молярным объемам жидкостей;
– равновесной концентрации в объемном растворе, равной растворимости, соответствует предельное значение коэффициента активности в уравнении парциальной изотермы адсорбции, величина которого пропорциональна произведению константы адсорбционного равновесия на растворимость;
– при адсорбции из водного раствора сильнополярных молекул (с большим дипольным моментом) в адсорбционной фазе проявляется диполь – дипольное отталкивание вследствие навязанной полем адсорбента однообразной ориентации диполей на границе раздела фаз, что приводит к уменьшению предельной плотности вещества в адсорбционной фазе. (6)
5.1 Аддитивность величин стандартного уменьшения свободной энергии адсорбции
Для количественной термодинамической характеристики адсорбируемости применяют величину стандартного уменьшения свободной энергии адсорбции ![]() . Поскольку дисперсионные силы, обусловливающие физическую адсорбцию, аддитивны, А.В. Киселев свел расчет уменьшения свободной энергии адсорбции молекул к вычислению инкрементов стандартного уменьшения свободной энергии адсорбции, обусловленных отдельными структурными элементами молекул адсорбата.
. Поскольку дисперсионные силы, обусловливающие физическую адсорбцию, аддитивны, А.В. Киселев свел расчет уменьшения свободной энергии адсорбции молекул к вычислению инкрементов стандартного уменьшения свободной энергии адсорбции, обусловленных отдельными структурными элементами молекул адсорбата.
При изучении адсорбции из растворов также можно было ожидать аддитивности энергий адсорбции, так как энергия сольватации (гидратации) аддитивно складывается из энергий сольватации отдельных элементов структуры молекулы.
Аддитивность изменения химических потенциалов при адсорбции из раствора была теоретически проанализирована в работе Осьцика и Ваксмундского. Рассматривая изменение химического потенциала в результате адсорбции многоатомных молекул из раствора, ли приняли в качестве исходной позиции, что общее изменение ![]() представляет собой сумму инкрементов, характеризующих изменение химического потенциала в результате адсорбции отдельных элементов структуры молекул таких, как группы –СН3 и =СН2, составляющие углеродный скелет органических молекул, или функциональные группы СООН, NO2 и т.п.
представляет собой сумму инкрементов, характеризующих изменение химического потенциала в результате адсорбции отдельных элементов структуры молекул таких, как группы –СН3 и =СН2, составляющие углеродный скелет органических молекул, или функциональные группы СООН, NO2 и т.п.
Теоретическое вычисление уме6ньшения свободно энергии при адсорбции из растворов до сих пор практически невозможно, особенно для случая адсорбции веществ, растворенных в воде. Поэтому при адсорбции растворенных веществ термодинамические величины, характеризующие адсорбционное равновесие, определяют экспериментально.
Таким образом, поскольку физическая адсорбция органических неэлектролитов и слабых электролитов неуглеродных материалах осуществляется, в основном, в результате дисперсионного взаимодействия, величина стандартного уменьшения свободной энергии адсорбции хорошо аппроксимируется суммой инкрементов, обусловленных вкладом отдельных структурных элементов и функциональных групп в это взаимодействие.
Стандартное мольное уменьшение свободно энергии адсорбции вещества может быть представлено в виде суммы инкрементов:
![]() (11)
(11)
т.е. величина стандартного уменьшения свободной энергии адсорбции отражает влияние химического строения молекулы на энергию адсорбционного взаимодействия и, следовательно, на константу адсорбционного взаимодействия. (2)
5.2. Парциальная константа адсорбционного равновесия при адсорбции из водных растворов на пористых углеродных сорбентах
Парциальную константу адсорбционного равновесия ![]() можно представить следующим уравнением:
можно представить следующим уравнением:
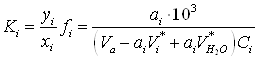 (12)
(12)
где ![]() и
и![]() – молярные доли растворенного вещества в адсорбционной фазе и равновесном состоянии;
– молярные доли растворенного вещества в адсорбционной фазе и равновесном состоянии; ![]() – удельная адсорбция растворенного вещества;
– удельная адсорбция растворенного вещества; ![]() – предельно-адсорбционный объем пор адсорбента;
– предельно-адсорбционный объем пор адсорбента; ![]() – молярный объем;
– молярный объем; ![]() – парциальный коэффициент активности компонента;
– парциальный коэффициент активности компонента; ![]()
Это – уравнение парциальной изотермы адсорбции, поскольку оно связывает равновесные величины адсорбции данного компонента с его равновесной концентрацией. Необходимые для расчетов величины молярных объемов компонентов раствора могут быть вычислены из их плотности в жидком состоянии:
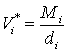 (13)
(13)
где ![]() – плотность жидкого компонента;
– плотность жидкого компонента; ![]() – его молекулярная масса.
– его молекулярная масса.
Эти же величины можно найти как произведение ван-дер-ваальсовской площади проекции адсорбированной молекулы на ван-дер-ваальсовский размер ее, нормальный к плотности проекции ![]() , т.е. на так называемую толщину молекулы.
, т.е. на так называемую толщину молекулы.
В уравнение парциальной изотермы адсорбции удобно вести величину относительного заполнения объема адсорбционной фазы органическим компонентом ![]() . Поскольку
. Поскольку ![]() , а уравнение (13) можно записать в следующем виде:
, а уравнение (13) можно записать в следующем виде:
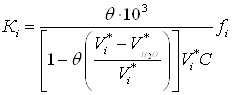 (14)
(14)
При вычислении константы адсорбционного равновесия в качестве стандартного выбрано состояние бесконечного разбавления в растворе в адсорбционной фазе, когда заполнение адсорбционной фазы стремится к нулю.
Поскольку коэффициент активности ![]() в уравнении (14) становится равным единице для стандартного состояния, т.е. при бесконечно малой величине
в уравнении (14) становится равным единице для стандартного состояния, т.е. при бесконечно малой величине ![]() , для нахождения числового значения константы адсорбционного равновесия экспериментальные данные, изображенные в координатах
, для нахождения числового значения константы адсорбционного равновесия экспериментальные данные, изображенные в координатах ![]() , должны быть экстраполированы до значения
, должны быть экстраполированы до значения ![]() . На рис. 5 показано графическое выделение логарифма парциально константы адсорбционного равновесия хлороформа, фенола, анилина, -хлоранилина, -нитрофенола, нитробензола и -нитроанилина из водных растворов на угле КАД (по уравнениям (13) и (14)).
. На рис. 5 показано графическое выделение логарифма парциально константы адсорбционного равновесия хлороформа, фенола, анилина, -хлоранилина, -нитрофенола, нитробензола и -нитроанилина из водных растворов на угле КАД (по уравнениям (13) и (14)).
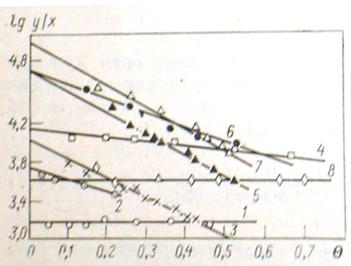
Рис. 5. Графическое вычисление парциальной константы адсорбционного равновесия хлороформа (1), фенола (2), анилина (3), -хлоранилина (4), -нитрофенол (5), нитробензол (6) и -нитроанилин (7) на активном угле КАД и бензола (8) на ацетиленовом техническом углероде (саже)
Из рис. 5 видно, что в подавляющем большинстве случаев существует линейная зависимость между ![]() и
и ![]() , что облегчает экстраполяцию значения
, что облегчает экстраполяцию значения ![]() до
до ![]() . В этом методе используются только определяемые величины: предельно-адсорбционный объем пор адсорбента и молярный объем адсорбируемого вещества, что делает применение метода особенно удобным для практических вычислений.
. В этом методе используются только определяемые величины: предельно-адсорбционный объем пор адсорбента и молярный объем адсорбируемого вещества, что делает применение метода особенно удобным для практических вычислений.
5.3 Коэффициент активности. Предельное значение коэффициента активности
Из уравнения (13) можно записать
 (15)
(15)
При исследовании адсорбции из растворов условием стандартного состояния является отсутствие взаимодействия между молекулами растворенного вещества, а также с молекулами воды, которое нарушило бы ее структуру. Этому стандартному состоянию соответствует бесконечно разбавление органического компонента водой в равновесном растворе и адсорбционной фазе.
Рассмотрим предельное значение коэффициента активности при ![]() . Подставив
. Подставив ![]() в уравнение (14), получим:
в уравнение (14), получим:
![]() (16)
(16)
где ![]() – равновесная концентрация, соответствующая плотному заполнению адсорбционной фазы, т.е.
– равновесная концентрация, соответствующая плотному заполнению адсорбционной фазы, т.е. ![]() .
.
Наибольшему заполнению адсорбционной фазы соответствует равновесная концентрация, равная концентрации насыщенного раствора Cs:
![]() при
при ![]() (17)
(17)
Поскольку вид функции ![]() от θ в общем случае выражается кривой, которая монотонно изменяется от
от θ в общем случае выражается кривой, которая монотонно изменяется от ![]() (при
(при ![]() ) до
) до ![]() (при
(при ![]() ), для приближенного вычисления
), для приближенного вычисления ![]() при любом
при любом ![]() можно использовать широко распространенный в химической технологии прием «рабочей линии». Для этого на ординате при
можно использовать широко распространенный в химической технологии прием «рабочей линии». Для этого на ординате при ![]() откладывают величину
откладывают величину ![]() и соединяют эту точку с началом координат. Тогда приближенное значение
и соединяют эту точку с началом координат. Тогда приближенное значение ![]() при любом значении
при любом значении ![]() можно найти из соотношения:
можно найти из соотношения:
![]() (18)
(18)
На рис. 6 показано изменение ![]() от
от ![]() для нитробензола (дипольный момент равен 3,96D). Штриховая прямая получена для значений
для нитробензола (дипольный момент равен 3,96D). Штриховая прямая получена для значений ![]() , рассчитанных по растворимости.
, рассчитанных по растворимости.
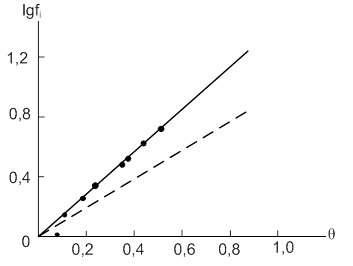
Рис.6. Зависимость lgfi от θ для молекул нитробензола, адсорбированного на угле КАД (штриховая линия рассчитана по растворимости)
Из рис. 6 видно, что до относительно заполнения ![]() эти прямые не совпадают. Еще более сильное диполь – дипольное отталкивание наблюдается в случае адсорбции -нитроанилина (дипольный момент равен 6,17D).
эти прямые не совпадают. Еще более сильное диполь – дипольное отталкивание наблюдается в случае адсорбции -нитроанилина (дипольный момент равен 6,17D).
Предельное значение коэффициента активности, таким образом, можно рассчитывать по константе адсорбционного равновесия и растворимости в тех случаях, когда диполь – дипольное отталкивание молекул в адсорбционной фазе отсутствует или незначительно (при малых дипольных моментах молекул).
5.4 Вычисление изотерм адсорбции органических веществ из водных растворов углеродными адсорбентами
Чтобы вычислить изотерму адсорбции, необходимо для разных значений заполнения адсорбционного объема ![]() (
(![]() ) рассчитать величины равновесной концентрации и удельной адсорбции. Значения равновесных концентраций (
) рассчитать величины равновесной концентрации и удельной адсорбции. Значения равновесных концентраций (![]() , ммоль/л) вычисляют по формуле
, ммоль/л) вычисляют по формуле
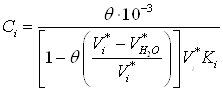 (19)
(19)
где ![]() – степень заполнения адсорбционного объема, задаваемая нами (0,1; 0,2; 0,3 и т.д.);
– степень заполнения адсорбционного объема, задаваемая нами (0,1; 0,2; 0,3 и т.д.); ![]() и
и ![]() - молярные объема извлекаемого органического вещества воды.
- молярные объема извлекаемого органического вещества воды.
Если известна структурная формула извлекаемого из воды вещества, то суммированием инкрементов ![]() для составных частей и структуры находят величину
для составных частей и структуры находят величину ![]() , а затем вычисляют константу адсорбционного равновесия по формуле
, а затем вычисляют константу адсорбционного равновесия по формуле
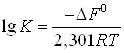 (20)
(20)
Удельную адсорбцию рассчитывают по формуле (в ммоль/г)
![]() (21)
(21)
Поскольку предельный коэффициент активности определяется соотношением ![]() , для разных значений констант адсорбционного равновесия
, для разных значений констант адсорбционного равновесия ![]() можно рассчитать те концентрации, при которых предельный коэффициент активности равен 1. Рассчитанные концентрации, соответствующие коэффициентам активности
можно рассчитать те концентрации, при которых предельный коэффициент активности равен 1. Рассчитанные концентрации, соответствующие коэффициентам активности ![]() при разных значениях
при разных значениях ![]() приведены в табл. 1.
приведены в табл. 1.
Таблица 1.
Значения концентраций насыщенных растворов, для которых fi = 1 при разных значениях ![]()
| К при 25ºС | Сs при 25ºС (рассчитанное), моль/м3 |
16,75 21 23 25,1 | 872 4800 11200 25700 | 63,5 11,5 4,95 2,16 |
Расчет изотерм адсорбции только по константе адсорбционного равновесия ![]() возможен для веществ, у которых соотношения между константами адсорбционного равновесия и величинами растворимости близки к указанным в табл. 1. (2)
возможен для веществ, у которых соотношения между константами адсорбционного равновесия и величинами растворимости близки к указанным в табл. 1. (2)
6. Методы выбора и контроля АДсорбентов для очистки воды
Промышленность выпускает много адсорбентов, характеристики которых известны. В процессе использования адсорбента, однако, и особенно его регенерации, происходит изменение некоторых свойств материала по сравнению с исходными. Поэтому необходим достаточно частый контроль некоторых параметров адсорбента и сопоставление их с таковыми свежего адсорбента. Использование специальных сравнительных методик значительно сокращает затраты труда на подобный контроль.
Общепризнанными методами определения сорбционных характеристик служат: сорбционная емкость по бензолу, йоду, фенолу, мелассе и метиленовому синему; определение площадей поверхности, размеров и объемов пор методами ртутной порометрии и БЭТ. Знание этих характеристик позволяет предсказывать поведение системы адсорбат – адсорбент в условиях адсорбции из смеси известного состава.
Отсутствие в настоящее время данных о полном количественном составе большинства природных и сточных вод и, как следствие этого, использование в практике обобщенных показателей качества воды (БПК, ХПК, цветность и др.) усложняет прогнозирование процесса адсорбции. При этом возможно несоответствие оценок адсорбции по ряду индивидуальных веществ и обобщенным санитарным показателям. Например, для ГАУ различной структуры получены близкие адсорбционные характеристики по ХПК при доочистке сточных вод. На нынешнем уровне знаний при изучении очистки воды лишь непосредственная проверка извлечения исследуемым материалом реальных загрязнений из конкретного источника или стока дает окончательный количественный ответ о рациональности использования данного образца адсорбента.
Адсорбционная очистка воды ведется при очень низких концентрациях одного или нескольких соединений (0,01 – 1,0 ммоль/л), которые во многих случаях адсорбируются независимо друг от друга. При этом адсорбционный процесс часто протекает в области, где выполняется закон Генри: ![]() , а изотерма адсорбции линейна и проходит через начало координат
, а изотерма адсорбции линейна и проходит через начало координат![]() , что несколько облегчает исследование сорбции.
, что несколько облегчает исследование сорбции.
Линеаризация изотермы облегчает контроль адсорбционных параметров материалов. Во многих случаях линеариз