Прогрессирующие мышечные дистрофии
Прогрессирующие мышечные дистрофии – это группа наследственно обусловленных нервно-мышечных заболеваний, характеризующихся прогрессирующей мышечной слабостью, атрофией мышц, двигательными нарушениями (Гусев Е.И., Никифоров А.С., 2007).
1. Эпидемиология прогрессирующих мышечных дистрофий
Первое сообщение о прогрессирующей мышечной дистрофии было опубликовано в России в 1895 г. врачом В.К. Ротом, который назвал заболевание мышечной сухоткой. Заболевание описано во всех странах мира. Частота 3,3 на 100 000 населения, 14 на 100 000 родившихся. В подавляющем большинстве случаев болеют мальчики. Случаи заболевания у девочек крайне редки, хотя и возможны при кариотипе ХО и при структурных аномалиях хромосом (Хр21.2, ген DMD дистрофина), в 35—40% случаев носит семейный характер (Гринио Л.П., Агафонов Б.В. 1997).
Частота прогрессирующей мышечной дистрофии Дюшенна варьирует от 9,7 до 32,6 на 100 000 живорожденных мальчиков. Высокая распространенность заболевания в популяции в значительной мере связана с высокой частотой новых мутаций (Bejaoui K., Hirabayashi K., Hentati F. et al., 1995).
Офтальмоплегическая мышечная дистрофия относится к числу редких заболеваний, частота встречаемости в Европе составляет 1:100 000—200 000 человек (Гусев Е.И., Коновалов А.Н., Скворцова В.И., Гехт А.Б., 2009). Однако в некоторых этнических группах и территориальных группах с «эффектом основателя» частота ОФМД намного выше, например во франко-канадской популяции — 1:1000 человек, у евреев Бухары, — 1:600. Также описаны выборки больных с офтальмоплегической мышечной дистрофией в более чем 30 странах на всех пяти континентах (Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008).
Популяционная частота прогрессирующей мышечной дистрофии Эрба - Рота составляет 1,2-2,5 случая на 100 000 населения (Гехт Б.М. и Ильина Н.А., 1998).
Частота встречаемости плече-лопаточно-лицевой миодистрофии Ландузи — Дежерина составляет 2,9 на 100 000 населения (Яхно Н.Н., Штульмен Д.Р., Мельничук П.В., 2001).
2. Этиология
Причиной являются генетически обусловленные дефекты метаболизма или структуры мышечной ткани, приводящие к атрофии мышц, разрастанию соединительной ткани и увеличению жировой клетчатки (псевдогипертрорфии) (Гусев Е.И., Никифоров А.С., 2007).
Табл. 1 Гены, ответственные за возникновение прогрессирующих мышечных дистрофий (Nevo Y., Muntoni F., Sewery C. et al., 1998)
| Название | Английская аббревиатура, синонимы | Тип наследования | Локализация гена | Ген | Белковый продукт гена |
| ПМД Дюшенна | DMD | ХР | Xp21.2 | DMD (DYS) | Дистрофин |
| ПМД Беккера | BMD | ХР | Xp21.2 | DMD (DYS) | Дистрофин |
| ПМД Эмери-Дрейфуса со сцепленным с полом наследованием | EDMD, скапуло - перонеальная форма, плече - лопаточно - перонеальная | ХР | Xq28 | Ген Эмерина, EDMD | Эмерин |
| ПМД Эмери-Дрейфуса, аутосомно-доминантный тип | EDMD2, скапуло - перонеальная форма, плече - лопаточно - перонеальная | АД | 1q21.2 | Ген Ламина А/C (LMNA/C) | Ламин А/С |
| ПМД Эмери-Дрейфуса, аутосомно-рецессивный тип | EDMD2, скапуло - перонеальная форма, плече - лопаточно - перонеальная | АР | 1q21.2 | Ген Ламина А/C (LMNA/C) | Ламин А/С |
| ПМД Ландузи-Дежерина | FSHD1, лице - плече - лопаточная форма | АД | 4q35 | FSHMD1A | - |
| Группа конечностно-поясных ПМД | |||||
| Окулофарингеальная форма, аутосомно-доминантный тип | OPMD | АД | 14q11.2-13 | PABP2 | Полиаденилин - ассоциированный белок |
| Окулофарингеальная форма, аутосомно-рецессивный тип | OPMD | АР | 14q11.2-13 | PABP2 | Полиаденилин - ассоциированный белок |
Различные формы прогрессирующих мышечных дистрофий могут наследоваться аутосомно-доминантно, аутосомно-рецессивно, рецессивно, сцепленно с Х-хромосомой. Различные формы прогрессирующих мышечных дистрофий отличаются разным типом наследования, вариабельностью возраста начала заболевания, преимущественной локализацией поражения мышц и другими признаками (Крахмалева И.Н., Липатова Н.А., Шишкин С.С. и др., 1999).
Миодистрофии Дюшенна и Беккера являются аллельными вариантами экспрессии единого генетического дефекта в локусе Р21 Х-хромосомы. Ген является самым большим из известных на сегодняшний день и имеет очень сложную молекулярную организацию; состоит из 79 экзонов (информативно значимых участков ДНК). В 60—65 % случаев мутация представляет собой делецию гена дистрофина, а в 5—10 % — его дупликацию. Встречаются и точковые мутации гена (до 30 % случаев) (Самуэльс М., 1997). Высокая частота спорадических случаев миодистрофий Дюшенна и Беккера обусловлена чрезвычайно высокой частотой спонтанных мутаций гена, возможно, отчасти из-за его "гигантского" размера (Свердлов Е.Д., 1997). С локусом Р21 Х-хромосомы ассоциированы также другие, редко встречающиеся, клинические фенотипы: семейная Х-сцепленная миалгия с крампи, синдром Мак-Леода (повышение уровня КФК, акантоцитоз), квадрицепс-миопатия. Последняя является наиболее мягкой формой и характеризуется медленным профессированием слабости четырехглавых мышц бедра, гипертрофией голеней и повышением КФК. При миодистрофий Дюшенна уровень дистрофина не превышает 3 % от нормального, тогда как при болезни Беккера он колеблется от 3 до 20 % (Гехт Б.М. и Ильина Н.А., 1998).
Этиология псевдогипертрофической формы Дюшенна.
Псевдогипертрофическая прогрессирующая мышечная дистрофия (мышечная дистрофия Дюшенна, Xp21.2, ген DMD дистрофина) — возникает в результате дефектов гена, кодирующего белок дистрофин. Дистрофин локализован в плазматической мембране скелетных мышечных волокон и кардиомиоцитов. Мутации в гене дистрофина вызывают клинически явно различающиеся формы миодистрофий (Евтушенко С.К., Садеков И.А. 1994).
Некоторые авторы предполагают, что различия в клинике дистрофинопатий могут определяться характером мутаций, из которых одни приводят к сдвигу рамки считывания (в результате чего синтез соответствующего белка практически невозможен), тогда как другие повреждают ген, но не нарушают рамку считывания (результатом чего становится синтез измененного белка, частично способного к функционированию) (Евтушенко С.К., Садеков И.А. 1994). Первый вид мутаций обычно связывают с тяжелым течением болезни, второй - с более мягким. Данное предположение, известное как гипотеза Монако, нашло подтверждение в ряде публикаций (Гринио Л.П., Агафонов Б.В., 1997).
Вместе с тем накопление сведений о мутациях в гене дистрофина и изучение клинико-генетических корреляций при дистрофинопатиях способствовали выявлению случаев, трудно объяснимых с позиций гипотезы Монако или других гипотез, постулирующих жесткую связь особенностей клиники с глубиной повреждения функции белка. Еще в 1988 г. было опубликовано сообщение о нескольких больных, у которых выявлялись делеции экзонов 3-7 гена дистрофина и соответственно сдвиг рамки считывания, но при этом заболевание протекало в мягкой форме - как миодистрофии Беккера (Шишкин С.С., 1997). Даже если бы у пациентов только отсутствовал данный участок последовательности дистрофина (не говоря уже о нарушении рамки считывания для остальной части гена), можно было бы ожидать серьезного нарушения функции белка, так как, по имеющимся сведениям, именно эта часть молекулы дистрофина обеспечивает его связывание с актином (McKusik V., Amberger J., 2003). Однако болезнь в ряде случаев протекала относительно доброкачественно. Более того, известен пациент с делецией экзонов 3-9 в дистрофиновом гене, который до 60-летнего возраста и не подозревал о своей болезни, а в 67 лет сохранял способность к самостоятельной ходьбе (Ahn A.H., Kunkel L.M., 1998). Авторы, описавшие этот уникальный случай, предположили, что, следовательно, возможны делеции в функционально важной области дистрофинового гена, которые обеспечивают состояние с длительным бессимптомным течением и без существенного сокращения продолжительности жизни (Kaplan J.C., Fontaine В., 1999).
В других публикациях отмечалось, что крупные делеции, захватывающие 26% (экзоны 21-44) и даже 40% последовательности дистрофинового гена, иногда обусловливают позднее начало и очень мягкое течение болезни - такие пациенты в возрасте 55 и 60 лет сохраняли определенную двигательную активность (Шаховская Н.И., 2000).
Этиология офтальмоплегической мышечной дистрофии Кило-Невина.
В 1998 г. изолировали на хромосоме 14q11.2-13 ген поли-(А)-связывающего белка 2 (PAPB2, PABPN1), ответственный за синтез ядерного белка PABP2, служащего фактором полиаденилирования мРНК, и идентифицировали мутацию, заключающуюся в увеличении числа копий тринуклеотидных GCG-повторов в 1-м экзоне гена. В норме ген содержит шесть тандемных копий повторов GCG, а у больных их число достигает до 8—13. В некоторых популяциях экспансия числа тринуклеотидных повторов происходит за счет простого добавления GCG-повторов, в других — вместе с экспансией GCG-повторов происходит GCA-вставка.
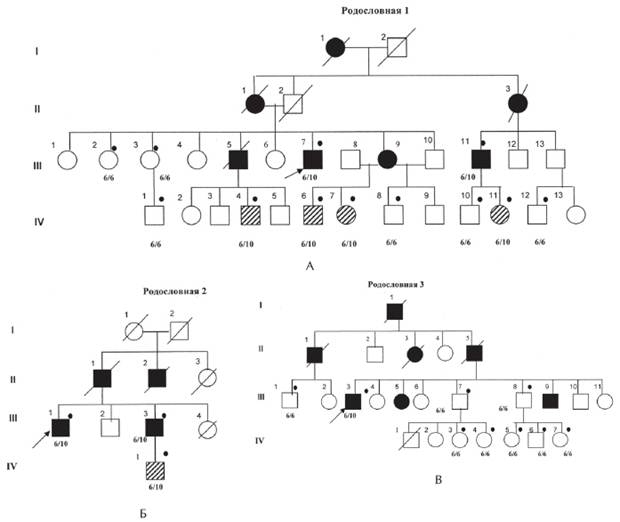
Рисунок 1. Рис. 1. А, Б, В. Родословные семей с офтальмоплегической мышечной дистрофии Кило-Невина с результатами ДНК-диагностики (Codere F., Brais B., Rouleau G., Lafontaine E., 2001).
Механизм развития экспансии тандемных повторов при офтальмоплегической мышечной дистрофии до сих пор неясен; высказываются предположения, что он может быть связан с неравным кроссинговером, разновидностью гомологичной рекомбинации, происходящей в зародышевых клетках во время мейоза или иногда митоза.
Также описаны случаи точечной мутации в гене PABPN1 22 и случаи гомозиготного носительства «промежуточного» аллеля гена, имеющего 7 GCG-повторов с аутосомно-рецессивным типом наследования с развитием более тяжелой клинической картины и более ранним дебютом симптомов заболевания (Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008).
Примечание. 6/6; 6/10 — результаты ДНК-анализа на мутацию в гене PABPN1; Закрашенный кружок —обследованные пациенты; закрашенный квадрат — больной с ОФМД, пустой квадрат — клинически здоровый; заштрихованный квадрат — клинически здоровый носитель мутации в гене PABPN1. I, II поколение — умершие родители, III поколение — больные и их сибсы, IV — дети, не достигшие возраста начала заболевания
Этиология конечно-поясной формы Эрба-Рота.
Известно не менее 9-ти локусов, ответственных за прогрессирующую мышечную дистрофию Эрба-Рота. Чаще всего вовлечен локус 15q15-q21.1 (McKusik V., Amberger J., 2003), реже вовлекается один из локусов, расположенных в коротком плече хромосомы 2 (Bashir R. et al., 1994), еще реже заболевание связывают с локусом 13q (Lim L.E., Duclos P., Broux O. et al., 1995).
В международной литературы конечностно-поясные формы обозначаются аббревиатурой LGMD (limb-girdle muscular dystrophy) c указанием типа, например LGMD 1A. Арабской цифрой 1 обозначаются типы с аутосомно-доминантным типом наследования, 2 – аутосомно-рецессивные формы (Шишкин С.С., Н.И. Шаховская, И.Н. Крахмалева, 2002). Как видно из представленной таблицы 2, конечностно-поясные формы прогрессирующих мышечных дистрофий – это целая группа генетически гетерогенных заболеваний, объединенных общей клинической картиной: прогрессирующая проксимальная мышечная слабость и гипотрофии, симптомы «крыловидных лопаток», «утиной походки», поясничный гиперлордоз. LGMD2A соответствует ювенильной конечностно-поясной форме прогрессирующих мышечных дистрофий Эрба-Рота (Умаханова Р. С. С. Жилина Г. Р. Мутовин, 2005).
Табл.2 Гены, дефекты которых ответственны за возникновение миодистрофии Эрба-Рота (пояснично-конечностная миодистрофия или LGMD) (Умаханова Р. С. С. Жилина Г. Р. Мутовин, 2005).
| Типы | Особенности | Тип наследования | Локализация гена | Ген | Белковый продукт гена |
| 1A | АД | 5q31 | Миотилин | ||
| 1B | Аллельная форма ПМД Эмери-Дрейфуса | АД | 1q21 | Ламин А/С | |
| 1C | АД | 3p25 | CAV3 | Кавеолин-3 | |
| 1D | АД | 7q | |||
| 1E | Дилатационная кардиомиопатия | АД | 6q23 | ||
| 1F | АД | 7q32 | |||
| 1G | АД | 4p21 | |||
| 2A (Эрба) | Начало 2-45 лет, в среднем 14-20 лет. | АР | 15q15.1-q15.3 | CAPN3 | Кальпаин-3 |
| 2B | Аллельная форма – дистальная дистрофия Миоши | АР | 2p13.1 | Дисферлин | |
| 2C | АР | 13q12 | SGCG |
| |
| 2D | АР | 17q21 | SGCA |
| |
| 2E | АР | 4q12 | SGCB |
| |
| 2F | АР | 5q33 | SGCD |
| |
| 2G | АР | 17q11-12 | Телетонин | ||
| 2H | АР | 9q31-33 | TRIM32 | ||
| 2I | Аллельная форма мерозиновой миопатии (ламинин-2) и конгенитальной мышечной дистрофией с мышечными гипертрофиями и нормальной ЦНС | АР | 19q13.3 | FKRP | Фукутин-связанный белок |
| 2J | Аллельная форма дилатационной кардиомиопатии 1G и Finnish дистальной миопатии | АР | 2q31 | Титин | |
| 2K | С умственной отсталостью | АР | 9q34 | POMT1 |
Экспрессивность генов конечно-поясной формы Эрба-Рота значительно варьирует не только в популяции, но даже в пределах одной пораженной семьи, что, по-видимому, и определяет различную тяжесть и прогрессирование миодистрофического процесса у больных, а также существование относительно доброкачественных или злокачественных форм патологии (Бадалян Л.О., 2008; Вельтищев Ю.Е. и соавт., 1998).
Этиология миодистрофии Дрейфуса-Хогана
В 1990-х годах последовательно были идентифицированы ген эмерина в локусе Хq28, ответственный за Х-сцепленную миодистрофию Дрейфуса-Хогана, и ген ламинов LMNA в локусе 1q21.2, вызывающий аутосомно-доминантную форму, вслед за чем появились многочисленные верифицированные наблюдения обоих генетических вариантов (Бадалян Л. О., Темин П.А., Калинин В.А. и др., 1990; Руденская Г.Е., Тверская С.М., Чухрова А.Л. и др., 2004).
Клиническая общность Х-сцепленной и аутосомно-доминантной форм неслучайна: она обусловлена тесным функциональным взаимодействием ламинов А/С и эмерина - мембранных белков, участвующих в образовании каркаса ядерной оболочки. При общем основном симптомокомплексе клиническая картина обеих генетических форм миодистрофии Дрейфуса-Хогана, особенно аутосомно-доминантной, демонстрирует разнообразие по возрасту начала, темпам прогрессирования, выраженности отдельных симптомов и тяжести болезни в целом, активности КФК, наличию атипичных признаков. Меж- и внутрисемейное клиническое разнообразие отмечалось уже в первых описаниях Х-сцепленной и аутосомно-доминантной миодистрофии Дрейфуса-Хогана вариантов (Бадалян Л. О., Темин П.А., Калинин В.А. и др., 1990).
В международных базах данных зарегистрировано более 100 мутаций гена эмерина и около 200 мутаций LMNA (большинство - при миодистрофии Дрейфуса-Хогана), действительное число мутаций несомненно больше, поскольку не все исследователи регистрируют свои находки в базах данных. Преобладают миссенс-мутации. Преобладающих по частоте («мажорных») мутаций нет, большинство мутаций обоих генов встречаются в единичных семьях, но некоторые описаны неоднократно. К ним относится мутация Arg249Gln в экзоне 4 гена LMNA, выявленная у больных с разными фенотипами. Мутация Arg249Gln возникает de novo, что позволяет предполагать наличие мутационной «горячей точки» в гене LMNA (Руденская Г.Е., Тверская С.М., Чухрова А.Л. и др., 2004). Так же имеет место мутация Arg377His в экзоне 6 (Мальмберг С.А., Петрухин А. С., Широкова В.И., 2000).
Нормальный биохимический продукт гена эмерин – представляет собой обогащенный аминокислотой (серином) белок, состоящий из 254 аминокислот. Эмерин экспрессируется преимущественно в скелетных, гладких мышцах и кардиомиоцитах; ему принадлежит значительная роль в организации клеточного цитоскелета и везикулярного транспорта. В сердечной мышце эмерин обеспечивает межклеточную адгезию и осуществление контактов между кардиомиоцитами. Типичная мутация представлена делецией гена и приводит к прекращению синтеза эмерина (Крахмалева И.Н., Липатова Н.А., Шишкин С.С. и др., 1999).
Этиология миодистрофии Бетлема
Редкая доброкачественная миодистрофия, наследуемая по аутосомно-доминантному типу. Установлена генетическая гетерогенность болезни: один из генов картирован в локусе 21q22, другой — 2q37. В результате мутаций нарушается синтез субъединиц коллагена VI типа, который обеспечивает связь базальной мембраны с гликопротеинами внеклеточного матрикса (Вельтищев Ю.Е., Темин П.А., 1998).
3. Патогенез
Существует несколько гипотез патогенеза прогрессирующих мышечных дистрофий. К настоящему времени точно установлено, что важным патогенетическим звеном является повышенная проницаемость мембран мышечных клеток (Евтушенко С.К., Садеков И.А. 1994). Имеются также данные, прямо или косвенно указывающие на существование мембранного дефекта при других прогрессирующих мышечных дистрофиях (Иллариошкин С.Н., Иванова-Смоленская И.А.. 1998). В частности, к свидетельствам повреждения мембран относят многократное повышение содержания в крови пациентов ряда мышечных ферментов и других мышечных белков (креатинфосфокиназы, трансаминаз). Наряду с этим отмечается, что существенную роль в развитии дистрофического процесса при ПМ могут играть нарушения обмена Са2+, приводящие к повышению его концентрации в цитоплазме клеток и активации Са2+-зависимых нейтральных протеиназ, которые в свою очередь запускают процессы разрушения мышечных белков (Горбунова В.Н., Савельева Е.А., Красильников В.В., 2000). Обсуждается также гипотеза об участии активных форм кислорода и свободных радикалов в запуске механизмов клеточной гибели при прогрессирующих мышечных дистрофиях. Однако принципиально важным представляется то, что мембраны мышечных клеток при прогрессирующих мышечных дистрофиях становятся проницаемыми для многих внутриклеточных белков и эти белки из клеток попадают в кровь (Баранов В.С., 1999).
По всей видимости, мышечные белки в крови могут восприниматься иммунной системой организма как чужеродный материал, и тогда на него будет возникать иммунный ответ, усиливающийся с возрастом пациента. В принципе такой иммунный ответ должен вести ко вторичному повреждению мембран мышечных клеток и еще больше усиливать выход в кровоток мышечных белков. Тем самым, возможно, определяется прогрессирование болезни. По крайней мере роль подобных аутоиммунных механизмов отмечена в патогенезе уже целого ряда заболеваний (Горбунова В.Н., Савельева Е.А., Красильников В.В., 2000).
Более того, обычно в мышцах пациента с миодистрофией Дюшенна наблюдаются разрастание соединительной ткани, инфильтрация лимфоцитами и жировое перерождение мышечных волокон. Все это в какой-то степени сходно с морфологической картиной других аутоиммунных заболеваний (Иллариошкин С.Н., Иванова-Смоленская И.А.. 1998).
Определенным подтверждением данного механизма патогенеза прогрессирующих мышечных дистрофий является успешное использование иммунодепрессантов с целью ослабления иммунного ответа организма на белки, поступающие в кровоток пациента из мышц, охваченных дистрофическим процессом (Шишкин С.С., Калинин В.Н., 1999).
Показано, что имеют место нарушения многих биохимических констант, различные электрофизиологические и ультраструктурные изменения. Определенную роль в развитии патологического процесса при прогрессирующих мышечных дистрофиях играет синтез неполноценных мышечных белков актина и миозина, сопровождающийся их ускоренным распадом. Обнаружены нарушения активности ряда неспецифических ферментов (креатинфосфокиназы, альдолазы и др.). Выявлены нарушения энергетического обмена, выражающиеся в быстром распаде соединений, используемых в качестве энергетических ресурсов при сокращении мышц. Определенную роль в развитии патологического процесса играет нарушение строения клеточных мембран, приводящее к изменению их проницаемости для ионов калия, натрия. Эти ионы имеют значение для сокращения мышц. В развитии дистрофии мышц определенная роль принадлежит патологии капилляров и нарушениям строения соединительной ткани (Liu J., Aoki M., Illa I. et al., 1998).
При прогрессирующих мышечных дистрофиях основной патологический процесс развивается в мышечной ткани; при другой группе болезней изменения в мышцах возникают вторично, первично нарушается структура нервной клетки и волокна. Эти заболевания носят название неврогенных мышечных атрофии. К ним относят спинальные (протекающие с преимущественным поражением двигательных клеток спинного мозга) и невральные (с поражением периферических нервов) амиотрофии (McNally E., Passos-Bueno R., Bonnemann C.G. et al., 1996).
В группу прогрессирующих мышечных дистрофий относят заболевания, различающиеся по времени появления клинических симптомов, преимущественной локализации мышечных атрофии, характеру их распространения, темпу нарастания патологических изменений и типу наследования (Шишкин С.С., 1998).
Основные патоморфологические изменения при прогрессирующих мышечных дистрофиях находят в мышцах. Они выражаются в атрофии отдельных мышечных волокон. Миофибриллы утрачивают поперечную исчерчешюсть, а иногда и полностью разрушаются. В ядрах мышечных клеток также обнаруживают изменения. Они становятся крупнее обычных, содержат различные включения, иногда сморщиваются. На месте атрофированных волокон интенсивно разрастается жировая и соединительная ткань. Нервные волокна и нервные клетки остаются относительно сохранными. Выраженные изменения находят в сосудах мышц, в которых имеется тенденция к сужению и образованию тромбов (Иллариошкин С.Н., Иванова-Смоленская И.А.. 1998).
4. Клиническая классификация (Казаков В.М., 2001)
| Тип | Генетический механизм | Клинические признаки | Вовлечение других систем органов | ||
| Дюшенна | Х-хромосомная рецессивная мутация дистрофин-гена | Начало в возрасте до 5 лет; прогрессирующая слабость мышц тазового и плечевого пояса; неспособность ходить после 12 лет; кифосколиоз; дыхательная недостаточность в возрасте 20-30 лет | Кардиомиопатия; снижение интеллекта | ||
| Беккера | Х-хромосомная рецессивная мутация дистрофин-гена | Начало в раннем или позднем возрасте; медленно прогрессирующая слабость мышц тазового и плечевого пояса; сохранение способности ходить после 15 лет; дыхательная недостаточность после 40 лет | Кардиомиопатия | ||
| Миотоническая | Аутосомно- доминантный; расширение нестабильного участка ДНК хромосомы 19ql3,3 | Начало в любом возрасте; медленно прогрессирующая слабость мышц век, лица, шеи, дистальных мышц конечностей; миотония | Нарушение сердечной проводимости; психические нарушения; катаракты, лобная алопеция; атрофия гонад | ||
| Плече-лопаточно-лицевая | Аутосомно-доминантный; часто мутации хромосомы 4q35 | Начало в возрасте до 20 лет; медленно прогрессирующая мышечная слабость лицевой области, плечевого пояса, тыльного сгибания стопы | Гипертензия; глухота | ||
| Плечевого и тазового пояса (возможны несколько заболеваний) | Аутосомно-рецессивный или доминантный | Начало с раннего детства до среднего возраста; медленно прогрессирующая слабость мышц плечевого и тазового пояса | Кардиомиопатия | ||
| Глазо-глоточная | Аутосомно-доминантный (Французская Канада или Испания) | Начало в 50-60 лет; медленно прогрессирующая слабость мышц: наружных глазных, век, лица и глотки; крикофарингеальная ахалазия. | Церебральные, глазные | ||
| Врожденная (включает несколько заболеваний, в том числе типы Фукуяма ицеребро-окулярная дисплазия) | Аутосомно-рецессивный | Начало при рождении; гипотония, контрактуры, задержка развития; в одних случаях — ранняя дыхательная недостаточность, в других — более благоприятное течение болезни | |||
| Основные выявленные клинические варианты миодистрофии Дюшена (МДД) | |||||
| Вариант МДД | Возраст начала болезни, годы | Способность к ходьбе и состояние опорно-двигательной системы | Масса тела, интеллект, осложнения | Число случаев и процент от общего числа пациентов | |
| I (классическое течение) | 2-5 | Теряет способность ходить в 10-12 лет. Генерализованная мышечная слабость, затем сколиоз, контрактуры голеностопных, коленных и других суставов | Масса тела снижена. Психическое развитие в норме; кардиомиопатия обнаруживается после 8-10 лет | 62(30,2%) | |
| II (с кушингоидным синдромом) | 2-5 | Теряет способность ходить в 10 лет или ранее. Генерализованная мышечная слабость, затем сколиоз, контрактуры голеностопных и других суставов | Ожирение (лунообразное лицо, отложение жира по женскому типу). Kардиомиопатия обнаруживается после 10 лет | 44(21,5%) | |
| III (врожденная форма) | 1-2-ой год жизни | Теряет способность ходить до 10 лет, иногда в 6,5-7 лет. Ранние множественные контрактуры. Быстрое прогрессирование | Масса тела снижена или в норме. Задержка психического развития. Kардиомиопатия обнаруживается в 7-10 лет | 28(13,7%) | |
| IV (кардиомиопатический) | 2-6 | В 6,5-7 лет обнаруживается кардиомиопатия при небольших проявлениях мышечной слабости (затруднения при подъеме по лестнице). Относительно медленное прогрессирование | Масса тела снижена или в норме. Психическое развитие в норме | 15(7,3%) | |
| V (смешанный) | 1-6 | Теряет способность ходить в 10-12 лет или ранее. Генерализованная мышечная слабость | Различные сочетания | 56(27,3%) | |
5. Клиника
Первые признаки болезни проявляются нарастающей слабостью тех или иных групп мышц, утомляемостью при легких физических нагрузках, симметричными атрофиями мышц (Евтушенко С.К., Садеков И.А. 1994).
Характерными симптомами прогрессирующих мышечных дистрофий являются мышечная слабость и атрофия мышц, которые могут проявляться в различные возрастные периоды, но чаще развиваются в детском и юношеском возрасте (Гаусманова - Петрусевич И., 2001). Дети поздно начинают ходить, быстро утомляются, неуклюжи в ходьбе, спотыкаются при беге, часто падают, с трудом поднимаются по лестнице. Двигательные нарушения постепенно прогрессируют. Возникает миопатическая утиная походка. В случае поражения мышц тазового пояса и конечностей затруднен переход из горизонтального положения в вертикальное; при поражении дистальных групп мышц ног появляется петушиная походка. Стойкость и нарастание двигательных нарушений позволяют диагностировать миодистрофию уже на ранних стадиях заболеваниях. При обследовании больного обнаруживают генерализованную или локальную атрофию мышц. Локальная атрофия мышц выявляется лишь на ранних стадиях заболевания, по мере прогрессирования патологического процесса атрофия мышц приобретает генерализованный характер вплоть до мышечной кахексии. Атрофированные мышцы истончены, дряблые при пальпации, однако следует отметить, что наряду с атрофией мышц выявляется псевдогипертрофия (замещение атрофированных мышц жировой клетчаткой, соединительной тканью). Миодистрофический процесс сопровождается поражением соединительной ткани, миосклерозом, развитием сухожильно-связочных ретракций, ограничением объема движений в суставах, укорочением пяточного (ахиллова) сухожилия, контрактурами. Одновременно с развитием мышечных атрофий снижаются сухожильные рефлексы, в первую очередь коленные (Вельтищев Ю.Е., Темин П.А., 1998).
Поражение мышц плечевого пояса приводит к ограничению движений в плечевых суставах. Больные не могут поднять руки выше горизонтального уровня, в то время как объем движений в локтевых и лучезапястных суставах и сила мышц длительное время остаются сохранными. При попытке поднять больного подмышки его голова как бы проваливается в плечи — симптом «свободных надплечий». Лопатки отстают от туловища — симптом «крыловидных лопаток». При поражении мышц тазового пояса возникают затруднения при подъеме на лестницу, вставании из положения сидя. При этом больной оказывает себе помощь, опираясь на посторонние предметы, встает в несколько этапов («лесенкой»). Изменяется походка: она становится переваливающейся, раскачивающейся - «утиная» походка. Атрофия косых мышц живота приводит к развитию «осиной» талии. Слабость длинных мышц спины нарушает осанку, приводит к искривлению позвоночника и выпячиванию живота (Гаусманова - Петрусевич И., 2001).
Поражение мышц костей и стоп сопровождается их слабостью. Походка больных становится своеобразной. Для того чтобы не зацепиться носком отвисающей стопы за пол, больные вынуждены высоко поднимать голень— «петушиная» походка (Евтушенко С.К., Садеков И.А., 1994).
При слабости и атрофии мышц лица отмечается отсутствие морщин на лбу (симптом «полированного лба»). Наблюдается гипомимия: больные не могут плотно зажмурить глаза, надуть щеки, вытянуть губы в трубочку и т. д. В некоторых случаях вследствие замещения губных мышц соединительной и жировой тканью губы утолщаются (напоминают губы тапира) (Hoffmann E.P., Kunkel L.M., Angelini C. et al., 2009).
При поражении наружных глазных мышц отмечается ограничение объема движения глазных яблок; иногда они становятся полностью неподвижными (McNally E., Passos-Bueno R., Bonnemann C.G. et al., 1996).
Если в патологический процесс вовлекаются мышцы глотки и гортани, возникает осиплость голоса и нарушается акт глотания. Поражение межреберных мышц ведет к дыхательной недостаточности и заболеваниям легких и сердца (Minetti C., Sotgia F., Bruno C. et al., 1998).
При неврологическом обследовании больных с прогрессирующими мышечными дистрофиями наряду с ограничением объема движений, снижением силы мышц и их атрофией выявляются мышечная гипотония, снижение или полное отсутствие сухожильных рефлексов (Moreira E., Vainzof M., Marie S. et al., 1997).
Темп прогрессирования патологического процесса зависит от формы заболевания и индивидуальных особенностей организма. В стадии выраженных нарушений вследствие атрофии мышц и отсутствия движений могут формироваться контрактуры (тугоподвижность или невозможность движения в суставах) (Minetti C., Sotgia F., Bruno C. et al., 1998).
Большинство форм прогрессирующих мышечных дистрофий не сопровождается снижением интеллекта. Больные критически относятся к своему дефекту. Иногда наблюдаются выраженные эмоциональные нарушения в виде повышенной раздражительности, подавленности настроения, замкнутости. Большинство больных успешно обучается по программе массовой школы (Muntoni F., Mateddu A., Marchei F. et al., 1993).
Исключение составляют больные псевдогипертрофической формой. При этой форме наблюдается выраженное снижение интеллекта. Данный вариант прогрессирующей мышечной дистрофии наследуется рецессивно, сцеплен-но с У-хромосомой. Основную массу больных составляют мальчики. Наряду с прогрессирующими атрофиями и слабостью мышц плечевого и тазового пояса у больных наблюдаются псевдогипертрофии (разрастания соединительной ткани, особенно в области икроножных мышц) и эндокринные нарушения (чаще ожирение). Некоторая задержка развития психических функций отмечается уже в первые годы жизни. Дети малоэмоциональны. Речь развивается с запозданием и носит примитивный характер. Отсутствует абстрактное мышление. Навыки опрятности и самообслуживания формируются с трудом. Интеллект обычно классифицируется как тяжелая дебильность или имбецильность; реже наблюдается идиотия (Гаусманова - Петрусевич И., 2001).
Псевдогипертрофическая злокачественная миодистрофия Дюшенна
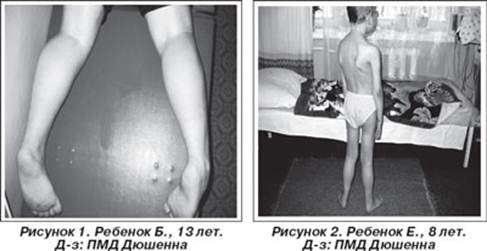
Первое наблюдение псевдогипертрофической миопатии принадлежит E.Meryon (1852), опубликовавшему в статье "К вопросу о жировой и гранулярной дегенерации мышц" семейный случай болезни у 4 братьев с аномальным увеличением икроножных мышц и контрактурами конечностей. G.Duchenne в 1861 г. описал больного с "псевдогипертрофическим мышечным параличом", обратив внимание на необычное сочетание увеличения икроножных мышц с прогрессирующей мышечной слабостью. В 1868 г. G.Duchenne опубликовал серию статей в журнале "Архив общей медицины", где представил систематизированный анализ болезни (Moreira E., Vainzof M., Marie S. et al., 1997).
Проявляется в возрасте 2—5 лет. Течение быстро прогрессирующее, злокачественное. Обездвиженность больных, как правило, наступает в возрасте 14—15 лет, смерть наступает в возрасте 15—18 лет, больные редко живут более 25 лет. К 8-10 годам большинство детей нуждается в ортопедических аппаратах; к 12 годам большинство детей не могут ходить. Первые признаки заболевания проявляются в 1-3 года жизни слабостью мышц тазового пояса. Уже на 1-м году обращает на себя внимание отставание детей в моторном развитии. Они, как правило, с задержкой начинают садиться, вставать, ходить. Движения неловкие, при ходьбе дети неустойчивы, часто спотыкаются, падают. В 2-3 года появляются мышечная слабость, патологическая мышечная утомляемость, проявляющаяся при физической нагрузке - длительной ходьбе, подъеме на лестницу, изменение походки по типу «утиной». В этот период обращает на себя внимание своеобразная «стереотипная» динамика движений детей во время вставания из горизонтального положения, с положения на корточках или со стул