Анализ сополимеризации индена с малеиновым ангидридом
Первоначально интерес к инден-кумароновым смолам был вызван тем, что они являлись альтернативой более дорогим синтетическим полимерам, поскольку сырьевой базой для них служат продукты переработки каменного угля. Благодаря достаточно высоким пластифицирующим свойствам, хорошей химической стойкости и водостойкости, а также относительной дешевизне эти смолы начали успешно применяться в лакокрасочной промышленности. Было показано, что они обладают и другими весьма ценными свойствами: высокой связующей и клеющей способностью, малой электро- и теплопроводностью, неплохой термостойкостью и способностью совмещаться с высыхающими маслами. Основным сырьем для их получения остаются продукты коксохимии, хотя возможно также использование побочных продуктов пиролиза нефти. Наличие такой сырьевой базы, хорошие технологические и эксплуатационные свойства при многообразии направлений использования инден-кумароновых смол способствуют сохранению устойчивого интереса к этому виду синтетических смол. В Донбассе проблема получения этих ценных смол с использованием кумарон-инденовой фракции (КИФ), многотоннажного отхода коксохомического производства, также достаточно актуальна.
Сополимеризация содержащихся в составе КИФ непредельных соединений в присутствии малеинового ангидрида и пероксидного инициатора может способствовать понижению температуры процесса получения смол, расширяет возможности использования продуктов на их основе, хотя этот вопрос изучен пока недостаточно.
Целью данной работы является изучение полимеризации инденовой фракции в присутствии МА и изучение молекулярной массы и полученных полимеров.
1. Обзор литературы
1.1 Теоретические основы процесса комплексно-радикальной полимеризации
Полимеризация виниловых и диеновых соединений представляет собой особый вид цепной реакции, характерной особенностью которой является то, что развитие кинетических цепей сопровождается ростом молекулярных цепей из молекул мономера (1, 2). Для цепной полимеризации (1, 2) характерно очень быстрое присоединение молекул мономера друг к другу без выделения побочных продуктов. Все способы инициирования полимеризации можно разделить на два класса. В одних случаях инициирование представляет собой реакцию присоединения к двойной связи мономера свободного радикала R*, образовавшегося тем или иным путём, а в других оно осуществляется в результате взаимодействия молекулы мономера с молекулами веществ, являющихся кислотами или основаниями Льюиса.
1.1.1 Общие положения радикальной (со)полимеризации
Процесс радикальной полимеризации можно изобразить следующей схемой (1):
| 1. | Реакция инициирования, приводящая к образованию из мономерных молекул М1 реакционных центров |
|
| 2. | Развитие реакционной цепи через активные полимеры, сопровождающееся ростом молекулярной цепи. Активные полимеры – промежуточные продукты полимеризации. |
|
| 3. | Обрыв реакционной цепи, приводящий к образованию конечного продукта – неактивного полимера Рn. |
|
Рассмотрим подробнее процесс радикального инициирования. Инициаторы (2) представляют собой термически неустойчивые соединения, распадающиеся с образованием свободных радикалов. Свободный радикал R* образуется вследствие гомолитического распада молекулы инициатора при поглощении ею энергии: R:R → R·. Он атакует двойную связь в молекуле мономера, при этом свободно-радикальный активный центр перемещается с фрагмента инициатора на мономерное звено:
![]() (2.1)
(2.1)
Этот процесс электронной перестройки сопровождается высвобождением энергии порядка 20 ккал (80 кДж), так как p-электронный уровень расположен выше уровня s-электронов. Таким образом, свободно-радикальная атака мономера при инициировании полимеризации - экзотермический процесс, в то время как разложение инициатора на свободные радикалы – эндотермическая реакция. Разложение инициаторов на свободные радикалы может происходить под действием тепла, света или других видов энергии, а также под влиянием катализаторов. В качестве инициаторов в основном используют азосоединения, пероксиды, гидропероксиды, перэфиры и перкислоты. Скорость разложения инициаторов зависит от их химического строения, а также от температуры реакции и используемого растворителя.
Одним из наиболее употребляемых инициаторов виниловой полимеризации является пероксид бензоила (1). В настоящее время можно считать установленным, что при нагревании растворов пероксида бензоила во многих растворителях первичным процессом является распад пероксида бензоила на два бензоатных радикала:
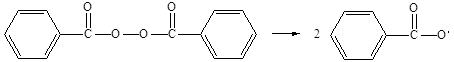 (2.2)
(2.2)
которые в дальнейшем способны распадаться с выделением СО2 и с образованием фенильных радикалов:
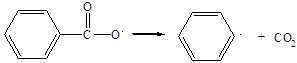 (2.3)
(2.3)
Если распад пероксида производится в присутствии энергичных акцепторов радикалов, то реакция (2.3) подавляется. Снижение выхода СО2 в присутствии виниловых соединений наблюдали японские авторы (3). В то же время при распаде пероксида бензоила в четырёххлористом углероде происходит выделение СО2 в количестве, соответствующем 96 % от теоретически возможного.(4) Так как бензоатный радикал, очевидно, не реагирует с четырёххлористым углеродом, то в этом случае почти все бензоатные радикалы распадаются согласно реакции (2.3). Кинетика распада пероксида бензоила в различных условиях была подробно исследована (3-9). Полученные при этом результаты в основном сводятся к следующему.
1. Скорость распада пероксида бензоила сильно зависит от растворителя, в котором протекает реакция.
2. Кинетический порядок реакции распада также зависит от растворителя.
3. При уменьшении концентрации пероксида удельная скорость распада (скорость, отнесённая к начальной концентрации пероксида) во многих случаях уменьшается и для различных растворителей стремится к одинаковой величине.
4. Добавление некоторых веществ, в частности виниловых соединений, к растворителям, в которых протекает быстрый распад пероксида, приводит к снижению удельной скорости реакции до величины, наблюдаемой при распаде пероксида в разведенных растворах.
Таким образом, распад пероксида представляет цепную реакцию (6-8), причём длина цепей зависит от природы растворителя. Предложено (6) следующее выражение для скорости распада пероксида бензоила:
![]() , (2.4)
, (2.4)
где (ПБ)-концентрация пероксида бензоила, k1 –константа скорости первичного мономолекулярного распада, а член kц(ПБ)n характеризует скорость цепного распада пероксида.
При виниловой полимеризации скорость и эффективность инициирования определяется первичным распадом пероксида, поэтому важно знать константу скорости мономолекулярного распада пероксида kПБ. Данные распада пероксида бензоила в бензоле (начальная концентрация 0,00185 моль/л) при температуре 60-80ºС удовлетворяют уравнению (5):
![]() (2.5)
(2.5)
В ароматических растворителях, например в толуоле, цепной распад пероксида бензоила при концентрациях пероксида не больше 0,2 моль/л невелик и приводит к образованию несимметричных дифенилов и значительных количеств бензойной кислоты (~50% от теории). Лёгкость присоединения фенильного радикала к бензольному кольцу с образованием нереакционного радикала позволяет понять малую величину цепного распада пероксида в ароматических растворителях. По-видимому, бензоатные радикалы также могут присоединяться к ароматическому кольцу, что приводит к образованию эфира бензойной кислоты. Присоединение бензоатного радикала к бензолу протекает медленнее, чем декарбоксилирование, так как выход эфира составляет лишь 5-7% (9).
Цепной распад пероксида может быть подавлен добавками ингибиторов. Особенно эффективны в этом отношении виниловые соединения. Поведение виниловых соединений по отношению к пероксиду бензоила во многих отношениях аналогично поведению ароматических соединений. Бензоатные радикалы, первоначально образующиеся при распаде пероксида бензоила, могут или присоединяться к двойной связи, давая начало полимерным цепям, или отщеплять молекулу диоксида углерода с образованием фенильного радикала, который также может присоединяться к двойной связи. Конкуренцию между реакцией декарбоксилирования и реакцией присоединения к двойной связи исследовали по выходу диоксида углерода при распаде пероксида бензоила в присутствии мономера. При увеличении концентрации мономера выход СО2 уменьшается, а при равных концентрациях зависит от природы мономера.
За инициированием следует стадия роста цепи (1, 2). На этой стадии активный центр, находящийся на первом мономерном звене, атакует двойную связь следующей молекулы мономера. Эта атака приводит к присоединению второго мономерного звена и переносу активного центра с первого на второе мономерное звено в соответствии со следующей схемой:
![]()
Необходимо отметить, что этот активный центр вновь способен к атаке следующей молекулы мономера с дальнейшим переносом неспаренного электрона на конец растущей цепи. Такой процесс, включающий последовательность актов присоединения молекул мономера к активному центру, получил название роста цепи. Возможны различные типы присоединения молекул мономера к активному концу растущей цепи. Так, различают присоединение по типу «голова к хвосту», «хвост к хвосту», «голова к голове», «хвост к голове», при этом под «головой» и «хвостом» мономерного звена понимают группы –СНХ– и –СН2–. Как упоминалось выше, присоединение очередного мономерного звена к концу растущей цепи сопровождается переходом p-электронной пары на уровень s-электронов и выделением энергии ~ 20 ккал. Следовательно, необходимо небольшое количество энергии извне для разложения инициатора и образования свободных радикалов, а далее полимерные цепи начинают рост с выделением большого количества энергии.
Поскольку при разложении инициатора одновременно образуется большое количество свободных радикалов, каждый из которых инициирует и продолжает рост цепей, в системе в любой момент времени существует определённое количество растущих цепей. В зависимости от ряда факторов, таких как температура, время реакции, концентрация мономера и инициатора, существует некоторая статистическая вероятность сближения двух растущих цепей и их взаимного столкновения. Когда происходит такое взаимное проникновение растущих клубков, возможно осуществление двух реакций, приводящих к обрыву цепей:
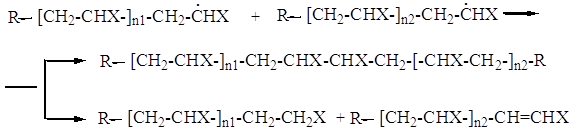
В первом случае две растущие цепи соединяются за счёт спаривания одиночных электронов активных центров, поэтому такой вид обрыва получил название рекомбинации. Во втором случае атом водорода отрывается от конца одной из растущих цепей, присоединяется к концу второй и образует стабильную связь с неспаренным электроном активного центра. Цепь, отдавшая атом водорода, стабилизируется за счёт образования концевой двойной связи. Такая реакция обрыва приводит к образованию двух полимерных молекул и носит название диспропорционирования.
Существует ещё один вид реакций ограничения длины растущих цепей, который происходит путём «передачи цепи» (2). Эта реакция протекает обычно путём отрыва атома водорода или любого другого подвижного атома от молекулы инициатора, мономера, мёртвых полимерных цепей или любых молекул, присутствующих в реакционных системах, включая растворитель и примеси. Ее можно схематически представить следующим образом:
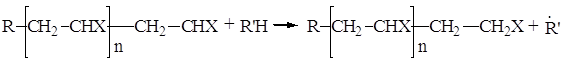
Растущая цепь при этом обрывается, но образуется новый радикал ![]() . Он теперь способен инициировать рост новой полимерной цепи. Таким образом, в ходе этой реакции одна живая, растущая цепь прекращает свой рост, тогда как новая начинает всё сначала.
. Он теперь способен инициировать рост новой полимерной цепи. Таким образом, в ходе этой реакции одна живая, растущая цепь прекращает свой рост, тогда как новая начинает всё сначала.
1.1.2 Полимеризация виниловых мономеров в присутствии комплексообразователей типа кислот Льюиса
Известно, что органические основания Льюиса (нитрилы, сложные эфиры, амиды и т. п.) способны образовывать достаточно прочные координационные комплексы с координационно-ненасыщенными соединениями непереходных металлов (чаще всего Zn, B, Al, Sn и др.), выступающих в таких случаях в качестве Льюисовых кислот. Непредельные органические соединения указанных выше классов, в частности, практически важные акриловые и метакриловые мономеры обычно полимеризуются по радикальному механизму. Образование их комплексов с кислотами Льюиса, естественно, приводит к изменению электронной структуры молекул мономеров и, следовательно, их реакционной способности. Поэтому кислоты Льюиса могут быть использованы в качестве модификаторов для целенаправленного изменения кинетических параметров отдельных элементарных стадий полимеризационных процессов.
В зависимости от природы мономеров и растворителя кислоты Льюиса могут образовывать донорно-акцепторые комплексы нескольких типов. В основе существующей классификации лежит тип орбиталей, участвующих в образовании межмолекулярных связей (10). Соединения - доноры электронов подразделяются на три группы: n, s и p в зависимости от типа высшей из занятых орбиталей (неподелённая электронная пара, s-связь и пара p-электронов соответственно). Координационно-ненасыщенные соединения металлов участвуют в комплексах за счёт вакантных орбиталей (V) атома металла. Наибольший интерес представляют следующие комплексы мономеров с кислотами Льюиса: nV-комплекс, в котором в качестве донора электронов выступает гетероатом с неподелённой парой заместителя мономера или радикала роста, и nVp-тройной комплекс, в котором акцептором является двойной комплекс nV, а донором – мономер или растворитель электронодонорного характера. Хотя те и другие комплексы по своей природе являются донорно-акцепторыми (ДА), в литературе это название в основном применяется к комплексам второго типа, а nV-комплексы по сложившейся терминологии называются координационными.
Комплексообразование между мономерами и кислотами Льюиса в координационных nV-комплексах надёжно установлено по данным ИК-, УФ- и ЯМР-спектроскопии. В ИК спектрах комплексов метилметакрилата и (мет)акрилонитрила с галогенидами металлов наблюдаются существенные сдвиги полос поглощения (11,12), соответствующих валентным колебаниям nС=О и nС≡N величиной от 20 до 155 см-1 и от 25 до 50 см-1. Частота валентных колебаний С=С связей изменяется незначительно, от 2 до 5 см-1. На основе этих данных сделан вывод о том, что комплексообразование указанных мономеров идёт преимущественно по функциональным группам заместителей и что роль двойной связи в этом процессе незначительна. Об этом свидетельствует и тот факт, что сдвиги полосы nС=О ММА и его предельного аналога – этилацетата при их взаимодействии с кислотами Льюиса близки. Весьма полезная информация получена также при анализе изменений частот деформационных колебаний δСН (в пределах от -12 до +22ºсм-1) и скелетных колебаний пиридинового кольца (21-30ºсм-1) в комплексах винилпиридинов с галогенидами цинка (13). Природа выявленных эффектов была интерпретирована в предположении, что ион металла комплексообразователя участвует в системе сопряжения мономера и смещает на себя его электронную плотность.
Для оценки изменения степени сопряжения двойной связи акриловых мономеров при комплексообразовании было использовано смещение полосы поглощения ![]() неплоских веерных колебаний С-Н связей метиленового фрагмента >C=СН2. Согласно (14), увеличение сопряжения двойной связи мономера приводит к повышению частоты
неплоских веерных колебаний С-Н связей метиленового фрагмента >C=СН2. Согласно (14), увеличение сопряжения двойной связи мономера приводит к повышению частоты ![]() , а усиление полярности - к её уменьшению. Следовательно, экспериментально наблюдаемый факт увеличения на 12-17ºсм-1 частоты колебаний
, а усиление полярности - к её уменьшению. Следовательно, экспериментально наблюдаемый факт увеличения на 12-17ºсм-1 частоты колебаний ![]() (мет)акриловых мономеров (бутилакрилата, акрилонитрила, метилметакрилата) в комплексах с с хлоридами цинка и олова свидетельствует об увеличении степени сопряжения двойной связи мономера.
(мет)акриловых мономеров (бутилакрилата, акрилонитрила, метилметакрилата) в комплексах с с хлоридами цинка и олова свидетельствует об увеличении степени сопряжения двойной связи мономера.
Влияние комплексообразования на степень сопряжения двойной связи было доказано также методом УФ-спектроскопии (10). Сопряжение в мономере косвенно может быть оценено по значению параметра Q схемы Q – e Алфрея – Прайса. Была установлена (13) линейная кореляция между lg Q и λπ-π*- полосой поглощения двойной связи мономера в УФ-спектре. Тазуке и Окамура (13) нашли, что при образовании комплекса с кислотами Льюиса наблюдается красное смещение полосы поглощения двойной связи. Этот факт указывает на увеличение резонансной стабилизации мономера в комплексе за счёт дополнительного сопряжения π-электронной системы мономера с атомом металла кислоты Льюиса. Уменьшение частоты поглощения, вызванное комплексообразованием, в некоторых случаях достигает 700 – 1500 см-1, что отвечает возрастанию параметра Q комплексо-связанных мономеров в 1,5 – 2 раза (15).
Об изменении полярности двойной связи мономеров при комплексообразовании свидетельствуют также данные ПМР спектров комплексов акриловых мономеров, например ММА, с хлоридами олова, алюминия и BF3, в которых химические сдвиги протонов мономеров смещены в область слабого поля по сравнению со свободными мономерами на 0,02-0,60 м.д. (11).
2 Полимеризация индена и кумарона
Интерес к полимеризации индена и кумарона был первоначально вызван тем, что они являются основными ненасыщенными компонентами сырого бензола и каменноугольного масла, способными к смолообразованию. До настоящего времени сырьем для производства инден-кумароновых смол остается тяжелый бензол, называемый также по преобладающим компонентам инден-кумароновой фракцией, хотя большую ценность представляет также и ксилольно-тяжелая фракция коксо-химического производства. Эти фракции содержат 50-70 и 35-40 % непредельных соединений и обе пригодны для получения кондиционных инден-кумароновых смол. Следует отметить, что в обеих указанных фракциях третьим по объему содержания ненасыщенным компонентом является стирол.
Наибольший интерес из указанных выше продуктов коксо-химического производства представляет инден, концентрация которого выше, чем стирола и кумарона. Инден легко бромируется, алкилируется, цианэтилируется. Однако основной областью его использования является процесс получения инден-кумароновых смол. Протекание этого процесса определяется ходом полимеризации и сополимеризации индивидуальных смолообразующих компонентов сырья. Не случайно поэтому большое число исследований было посвящено изучению химизма и условий полимеризации соединений инденового и кумаронового ряда. Начало им было положено работами Кремера и Шпилькера, опубликованными в 1890 г. (14, 16), а затем в разное время этот вопрос изучали Штермер, Штоббе и Фербер, Уитби и Кац, Штаудингер (14, 16), в 60-е годы ХХ в. обстоятельные исследования были выполнены Марешалем и др (15,16,17,18). Уже в первых работах было установлено, что полимеризация индена может протекать под действием света, тепла, давления и главным образом под влиянием различных катализаторов.
2.1 Основные закономерности полимеризации индена и кумарона
Полимеризация индена протекает под действием солнечного света, однако чрезвычайно медленно: при выдержке на свету в течение года удалось получить лишь небольшой выход полимера, содержащего от 8 до 16 молекул исходного мономера. Довольно подробно изучена, например, фотодимеризация индена (16, 19). Показано, что реакция протекает как через синглетное, так и через триплетное состояние индена. При несенсибилизированной фотодимеризации 80 % димера образуется через триплетный инден.
Более интенсивно протекает термическая полимеризация. Уже при обычной температуре происходит автополимеризация индена, скорость которой быстро возрастает с повышением температуры. Однако даже при температуре ~200ºС для обеспечения полноты реакции полимеризации требуется длительное время (14, 16). Наряду с полимеризацией при нагревании индена протекает реакция автовосстановления, в результате которой происходит образование трускена (С27Н18) и гидриндена (С9Н10), а если нагревать инден в присутствии воздуха, то происходит также его автоокисление. При повышении температуры молекулярная масса продукта уменьшается, например, от 886 до 676 при 178ºС и 200ºС. Как правило, молекулярная масса продукта реакции при термополимеризации не превышает 1000.
Термополимеризация кумарона и индена во многом сходны, отличие заключается в степени и скорости полимеризации. Если, например, при 20 ч нагревания индена с обратным холодильником было получено 30% смолы, то кумарон при нагревании в тех же условиях образовал лишь незначительное ее количество (14, 16). Молекулярная масса при термической полимеризации кумарона также ниже, чем при полимеризации индена.
Полимеризация индена и кумарона может быть инициирована различными способами. Радикальная полимеризация этих соединений изучена мало. Описана радикальная полимеризация и сополимеризация индена и кумарона в массе при давлении до 10000 атм в присутствии пероксида бензоила (17, 19). Инден полимеризуется в течение нескольких минут; при достижении 175ºС реакция носит взрывной характер. В результате реакции образуется твёрдый жёлтый полимер. Полимеризация кумарона протекает несколько медленнее, взрывной характер реакция приобретает только при 275ºС.
Наряду с ростом макромолекулы образовавшиеся свободные радикалы способствуют протеканию и других реакций. Например, бензильный радикал отрывает Н из положения 1 у индена с образованием толуола и нового радикала, который превращается далее в 1-бензилинден и 1,1-диинденил. Возможно также гомолитическое замещение в положениях 2 и 3 у индена, сопровождающееся присоединением PhCH2 по двойной связи и образованием 2- и 3-бензилинденов (20).
Радикальная полимеризация компонентов коксохимического сырья протекает с различной скоростью. Так, под действием 5 % гидроперекиси изопропилбензола при температуре 125ºС инден полимеризуется на 46-50 %, стирол и метилстиролы – на 89-90 %, кумарон практически совсем не полимеризуется (21).
Исследована, также электроинициированная полимеризация индена. При пропускании электрического тока через раствор перхлората лития в уксусном ангидриде, содержащий инден, в анодном пространстве происходит образование полииндена. Предполагается, что реакция развивается по ионному механизму, причем максимальный выход полимера достигает 16 %. Образующийся полиинден по структуре не отличается от полимера, полученного катионной полимеризацией.
Каталитической полимерации индена и кумарона посвящено наибольшее количество работ. Еще более века назад было установлено, что эти соединения способны полимеризоваться под действием различных катализаторов кислотного типа, например, серной кислоты и AlCl3. Ранние исследования были посвящены главным образом сернокислотной полимеризации. Под действием 75 %-ной кислоты образовывалась растворимая в бензоле смола, из которой удалось выделить диинден и тетрамер индена, названный параинденом, с температурой плавления 210ºС. При действии 95 %-ной серной кислоты были получены продукты, содержащие 16-22 молекулы индена с температурой плавления 220-280ºС (метаинден).
Кумарон под действием серной кислоты также дает продукты различной степени полимеризации, среди которых охарактеризованы α- и β-паракумарон со степенью полимеризации 4 и 8 соответственно.
Кроме серной кислоты, полимеризация указанных соединений может эффективно протекать под действием таких катализаторов, как ZnCl2, FeCl3, SnCl4, TiCl4, SbCl5, AlBr3, AlCl3, BF3. Во всех этих случаях происходит так называемая гомогенная (иногда псевдогомогенная) полимеризация, при которой катализаторы вводятся в реакционную среду либо в виде раствора, либо в виде жидких комплексов солей с растворителем. Возможно также применение гетерогенных катализаторов, таких как природные или синтетические алюмосиликаты, активированные минеральными кислотами, а также оксидами или солями (22).
Согласно современным представлениям для начала катионной полимеризации необходимо возникновение активных центров. В их образовании принимают участие катализатор – галогенид металла МХn, являющийся кислотой Льюиса, сокатализатор ВН (кислота Бренстеда, чаще всего, вода иди галоидводороды), выступающий в качестве донора протона, и исходный мономер. Как и при радикальном инициировании, процесс катионной полимеризации протекает в несколько стадий: инициирование, рост цепи и его прекращение, которое осуществляется в результате передачи цепи за счет переноса протона на другую частицу: молекулу мономера, растворителя, противоион или макромолекулу.
В целом при полимеризации индена наблюдаются общие закономерности, характерные для катионной полимеризации других непредельных соединений, в частности, виниловых мономеров. Течение процесса полимеризации и его результаты очень сильно зависят от природы применяемого катализатора. Для индена наибольшая характеристическая вязкость получена в присутствии BF3 и TiCl4. Степень полимеризации с увеличением концентрации катализатора до некоторого предела растёт, после чего остаётся почти постоянной.
Кроме того, при полимеризации индена отмечена характерная для катионной полимеризации зависимость скорости и степени полимеризации от природы растворителя, а именно повышение степени полимеризации с увеличением диэлектрической проницаемости растворителя. Высокую степень полимеризации обеспечивает применение в качестве растворителей галоидированных алифатических углеводородов, особенно хлористого метилена. При использовании ароматических растворителей, например толуола, степень полимеризации существенно понижается, что может быть связано с явлением передачей цепи, которая более ярко выражена в случае ароматических соединений.
Катионная полимеризация индена возможна в довольно широком диапазоне температур, от 273 до 173 К. При этом характеристическая вязкость увеличивается от 0,53 до 1,90 дл/г (растворитель – хлористый метилен, 100 г/л индена, катализатор - TiCl4 в концентрации 0,02 моль/л)(23). В этих же условиях при температуре 201 К молекулярная масса полииндена со снижением концентрации мономера до 50 г/л возрастает, а при дальнейшем уменьшении его концентрации также уменьшается. Это может быть объяснено, с одной стороны, снижением температуры реакции при меньших концентрациях мономера, с другой стороны, увеличением диэлектрической проницаемости среды за счёт повышения концентрации растворителя.
При благоприятных условиях полимеризации – низкой температуре реакции и применении растворителя с высокой диэлектрической проницаемостью - может быть получен полиинден с характеристической вязкостью около 2 дл/г, что соответствует молекулярной массе более миллиона (16).
Изучение катионной полимеризации кумарона в различных условиях (24) по методике, описанной ранее для индена, показало, что слабые катализаторы катионного типа не инициируют полимеризацию кумарона. Наиболее активным катализатором, как для индена, является BF3. Полимеризация кумарона с ним в растворителе с высокой диэлектрической проницаемостью при 201 К заканчивается за 5 мин и приводит к получению полимера с характеристической вязкостью 3,8 дл/г, что соответствует степени полимеризации не менее 8500. Как и для индена, полимеризация кумарона зависит от природы и диэлектрической проницаемости растворителя. Молекулярная масса полимера также увеличивается с понижением температуры реакции. В целом можно отметить, что полимеризация кумарона при равных условиях протекает медленнее, чем полимеризация индена, и с меньшим выходом, но полимер имеет более высокую характеристическую вязкость. Установлено, что по скорости катионной гомополимеризации с большинством катализаторов основные непредельные компоненты в составе коксохимического сырья располагаются в ряд : инден > стирол » кумарон (25).
2.2 Структура продуктов полимеризации индена и кумарона
Вопрос о структуре образующихся полимеров с самого начала исследований был предметом длительных научных споров. Димеру индена была приписана следующая насыщенная структура:
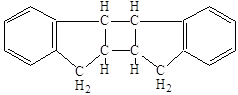 ,
,
подтвержденная более поздними исследованиями (18).
Аналогично в виде замкнутых насыщенных структур представляли первоначально молекулы тетрамера и более высокомолекулярных полимеров индена. Однако такое представление противоречило экспериментальным данным о наличии остаточной непредельности у продуктов полимеризации индена, поэтому для изображения дииндена предложили формулы:
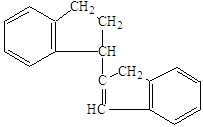 или
или 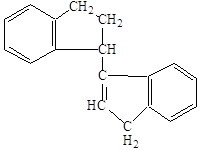 ,
,
которые были перенесены на строение продуктов более глубокой степени полимеризации. Такая структура полимеров объясняет их способность присоединять галоид (наличие двойной связи в молекуле полимера), а также способность продуктов низкой степени полимеризации к дальнейшему уплотнению, и наблюдаемую даже у твёрдых хрупких смол способность к присоединению кислорода с образованием пероксидов. Этим же объясняется постепенное уменьшение иодного числа у полимеров большей молекулярной массы, так как остаточная двойная связь приходится на всё более длинную молекулу полимера.
Спектроскопические исследования (26 - 28) кумарона и индена и полученных из них методом каталитической полимеризации продуктов подтверждают, что реакция полимеризации протекает за счёт раскрытия двойной связи пятичленного цикла. Поэтому представление о линейной структуре полимеров индена и кумарона получило наибольшее распространение. Длительное время полимеры кумарона и индена представляли в виде линейных цепей с расположением молекул по одну сторону от главной оси. Экхардт и Хайне (26) впервые высказали предположение о том, что под действием катализаторов катионной полимеризации, таких, как хлористый алюминий или трёхфтористый бор, из индена возможно образование трёх типов стереорегулярных полимеров: изотактических, атактических и синдиотактических, что доказывают и исследования ИК спектров полииндена (27).
Вполне определённо доказана возможность получения стереорегулярных полимеров из кумарона (23). Характерной особенностью кумарона является то, что он склонен к образованию оптически активных полимеров, имеющих диизотактическую структуру. В ИК спектре продукта термической полимеризации кумарона обнаружена полоса при n=3540 см-1 (27), которая может быть объяснена только образованием в поликумароне внутримолекулярной водородной связи между кислородом фуранового цикла связанной молекулы и одним из водородов другой молекулы, которая может возникнуть только в том случае, если цепь поликумарона имеет не линейное, а зигзагообразное строение:
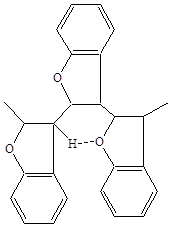
Регулярное расположение молекул в цепи поликумарона должно быть устойчивым, так как оно стабилизируется водородной связью. Получение оптически активного поликумарона открывает возможности для изготовления на его основе избирательных по оптической активности фильтров, адсорбирующих средств или ионообменных смол.
Получить оптически активные полимеры индена до сих пор не удалось. Таким образом, нет полной аналогии между строением полимеров индена и кумарона. Различия в структуре образующихся полимеров, а также в протекании процессов полимеризации кумарона и индена объясняются различными свойствами пятичленного цикла в том и другом случае.
2.3 Сополимеризация индена с кумароном и другими непредельными соединениями
В продуктах переработки каменноугольной смолы и сырого бензола инден и кумарон содержатся совместно. Поэтому особый интерес представляет исследование сополимеризации этих двух мономеров. Отметим, что в литературе нами найдены данные только по изучению катионного процесса.
В присутствии четырёххлористого титана реакция сополимеризации индена с кумароном протекает с хорошим выходом. При этом отмечено (23), что молекулярная масса закономерно понижается от 1,63 для полииндена до 0,28 дл/г с увеличением доли кумарона в смеси мономеров до 50 мол.%. Полимеризация в присутствии BF3 легко позволяет получить поликумарон и сополимер с 50 % индена с характеристической вязкостью 1,7 дл/г, что выше, чем с TiCl4.
Кроме индена и кумарона, в коксохимическом сырье могут содержаться также такие непредельные соединения, как стирол, a-метилстирол, циклопентадиен. В табл. 1.1 приводятся некоторые экспериментальные данные, характеризующие сополимеризацию этих соединений с инденом.
Таблица 1.1. - Условия сополимеризации индена со стиролом, a-метилстиролом, циклопентадиеном в хлористом метилене и вязкость (h) полученных сополимеров при выходе 100 % (по данным (23)).
| Второй мономер1) | Катализатор2) | Температура, ºС | h, 100см3/г |
| Стирол | TiCl4 | -72 | 0,19 |
| BF3 | -72 | 0,20 | |
| BF3 | -100 | 0,22 | |
| a-метилстирол | TiCl4 | -72 | 0,54 |
| BF3 | -97 | 1,45 | |
| Циклопентадиен | TiCl4 | -72 | 0,44 |
| TiCl4 | -93 | 0,85 |
1) концентрация 0,42 моль/л; 2) концентрация 0,02 моль/л.
При совместной полимеризации во всех случаях отмечается понижение молекулярной массы получаемого продукта. Особенно наглядно это видно на примере совместной полимеризации индена и стирола (23), если учесть, что в одинаковых условиях гомополимеризация стирола даёт продукт с вязкостью 0,56; a-метилстирола - 0,55; индена - 1,60 дл/г.
Сополимеризация кумарона со стиролом и a-метилстиролом при концентрации TiCl4 0,01 моль/л и температуре 201 К за 0,5 ч протекает с выходом 58 и 89 %, давая продукт, содержащий 48 и 44 % кумарона, с вязкостью 0,12 и 0,82 дл/г соответственно (23). Таким образом, анализ имеющихся экспериментальных данных показывает, что катионная гомополимеризация и сополимеризация индена и кумарона наиболее активно протекает под действием таких катализаторов, как хлористый алюминий и трёхфтористый бор. Совместная полимеризация нескольких мономеров приводит обычно к получению полимеров с меньшей молекулярной массой, чем при их раздельной полимеризации. Снижению молекулярной массы способствует также применение ароматических растворителей.
2.4 Модификация инден-кумароновых смол
Требования многочисленных потребителей к качеству инден-кумароновых смол (16) весьма разнообразны и не все эти требования могут быть удовлетворены при существующей сырьевой базе и технологии производства смол. Недостаточная свето- и атмосферостойкость инден-кумароновых смол, хрупкость, неполная растворимость во многих полярных растворителях, несовместимость с синтетическими плёнкообразующими веществами и плохая совместимость с высыхающими маслами, невозможность получения термореактивных плёнкообразователей – эти свойства не удовлетворяют лакокрасочную промышленность. Не соответствует современным требованиям относительно высокое остаточное содержание в смоле легколетучих веществ, особенно в тех случаях, когда переработка инден-кумароновой смолы осуществляется при повышенной температуре.
Большие возможности для изменения качества смол даёт модификация их различными химическими соединениями. Помимо получения инден-кумароновых смол с требуемыми свойствами, модификация может довольно существенно увеличить ресурсы сырья для из производства.
Одним из способов придания инден-кумароновой смоле новых физико-механических свойств является приготовление разнообразных компаундов. Уже в этом простейшем случае речь идёт не о механической смеси нескольк