Влияние структуры исходной ПАН-нити на структуру и свойства углеродного волокна
В. А. Тюменцев, А. А. Свиридов, С. А. Подкопаев, И. А. Ягафаров
Уникальные физико-механические свойства углеродного волокна (УВ) обусловлены особенностями его микроструктуры, в том числе размерами областей когерентного рассеяния (ОКР) и высокой степенью упорядоченности материала, формирующегося в процессе термостабилизации (200—300 °С) и высокотемпературной (-1500—3000 °С) обработки исходного волокна на основе полиакрилонитрила (ПАН-волокна) (1—5). В литературе достаточно подробно рассмотрено влияние режимов термостабилизации и высокотемпературной обработки на тепловые эффекты, удаление летучих, усадочные явления и физико-механические свойства образующегося нанодисперс-ного волокнистого материала (2—4). Однако закономерности структурных преобразований паракристал-лического полиакрилонитрила в термодинамически неравновесную нанодисперсную структуру термоста-билизированного волокна, а затем в структуру углеродного волокна в условиях высокоскоростного нагрева недостаточно изучены. В работе рассмотрены закономерности преобразования материала ПАН-волокна, полученного по диметилсульфоксидной технологии, в углеродное волокно.
Термостабилизацию ПАН-волокна осуществляли в атмосфере воздуха в изотермических условиях лабораторной установки и в высокотемпературной печи рентгеновского аппарата. Средние размеры ОКР волокна определяли методом рентгеноструктурного анализа (фильтрованное СиА^-излучение) по стандартным методикам. Межплоскостное расстояние фаз рассчитывали по центру тяжести дифракционных максимумов.
Особенностью температурных исследований in situ является то, что в процессе эксперимента информацию о структуре материала получают от одного и того же объема вещества, что позволяет оценить изменение количества исходной кристаллической фазы. Нагрев жгута в специальной печи рентгеновского аппарата до
240 °С проводили со скоростью 2,5 град/мин с последующей изотермической выдержкой в атмосфере воздуха до завершения структурных преобразований материала.
По мере повышения температуры образца ПАН-нити, полученной по режиму I, на дифрактограмме постепенно происходит смещение характерного для полиакрилонитрила максимума 220 в сторону меньших углов дифракции на -0,75 градуса, возрастает в -1,3 раза интенсивность сигнала в максимуме, а средние размеры областей когерентного рассеяния, рассчитанные по интегральной ширине, увеличиваются до 14,8 нм (рис. 1, табл. 1). В процессе изотермической выдержки интенсивность максимума 220 начинает уменьшаться. При этом одновременно происходит увеличение его интегральной ширины Pi. Структурные преобразования материала завершаются через 2,5 часа. Изменение условий формования ПАН-нити оказало существенное влияние на закономерности структурных преобразований материала. На кривой дифференциально-термического анализа максимум экзоэффекта, обусловленный протеканием реакций циклизации и окисления, сместился на 15 °С в сторону более низких температур.
В процессе нагрева волокна, полученного по режиму II (табл. 1), наблюдается смещение дифракционного максимума 220 в сторону меньших углов дифракции всего на -0,63 градуса, однако его интенсивность в максимуме возрастает в -1,6 раза, а средние размеры областей когерентного рассеяния увеличиваются до 17 нм. Во время изотермической выдержки материала также более активно развивается фазовый переход структуры ПАН в «предструктуру» углеродного волокна (рис. 2). Процесс завершается через 2 часа, и характерные для структуры ПАН дифракционные максимумы на рентгенограмме не обнаруживаются (табл. 1). Наблюдаемые изменения на дифрактограмме, а также данные, приведенные в (6), указывают на Cu-Ка — излучение, растворитель полиакрилонитрила — диметилсульфоксид то, что в начале окислительной термообработки происходит заметное увеличение средних размеров областей когерентного рассеяния полиакрилонитрила.
В процессе изотермической термообработки постепенно изменяется расположенный в области углов 263 -27 градусов третий широкий дифракционный максимум, обусловленный рассеянием рентгеновских лучей нанодисперсной фазой с межслоевым расстоянием, сравнительно близким к структуре графита. Авторы работы (7) полагают, что формирование этого максимума связано с рассеянием рентгеновских лучей на так называемой «разориентированной» фазе. Широкий третий максимум смещается в область меньших значений углов дифракции с уменьшением его интегральной ширины. Изменения на рентгенограмме обусловлены уменьшением межслоевого расстояния и некоторым ростом размеров областей когерентного рассеяния.
Структурные преобразования материала ПАН-волокна, полученного по режиму III, в процессе изотермической термообработки при 220 °С развиваются более активно. На дифрактограмме недоокисленной ПАН-нити наряду с характерными для структуры полиакрилонитрила максимумами при значениях углов дифракции 26) = 16,7 и 262 = 29 градусов отчетливо наблюдаются достаточно интенсивный третий дифракционный максимум (263 = 27,3 градуса, рис. 3). При этом интегральная ширина первого максимума Pi недоокисленной нити увеличилась, по сравнению с Pi исходного ПАН, почти на 40%. Следовательно частичный (развивающийся в локальных микрообъемах филамента) фазовый переход материала ПАН-нити, полученной по режиму III, сопровождается заметно большим диспергированием исходной фазы и, возможно, появлением микродеформаций этой структуры.
Недоокисленная ПАН-нить двухфазная состоит из кристаллической и нанодисперсной фазы, которую можно рассматривать как промежуточную между структурой ПАН и углеродного волокна. Исходная и вновь образующаяся фазы в термостабилизируемом волокне сосуществуют в широком температурном интервале. Увеличение температуры термообработки стимулирует завершение структурных преобразова-
Таблица 1
Изменение параметров структуры ПАН-волокна в процессе термообработки in situ в атмосфере воздуха
| г, "С | Время изотермического нагрева | Положение дифракционного максимума 26, град. | Интегральная ширина град. | Расстояние d, нм | Средние размеры OKI L, нм | Интенсивность дифрак ционного максимума /, отн. ед. |
| ПАН-нить, | полученная | по режиму | I | |||
| 75 | — | 17,15 | 0,8 | 0,517 | 10,2 | 170 |
| 170 | — | 16,75 | 0,8 | 0,529 | 10,2 | 175 |
| 200 | — | 16,6 | 0,7 | 0,534 | 12,7 | 195 |
| 220 | — | 16,55 | 0,65 | 0,535 | 13,7 | 210 |
| 240 | — | 16,45 | 0,6 | 0,539 | 14,8 | 220 |
| 240 | 15 мин | 16,4 | 0,65 | 0,540 | 13,8 | 190 |
| 240 | 30 мин | 16,4 | 0,75 | 0,540 | 11,9 | 130 |
| 240 | 1 ч | 16,4 | 1,0 | 0,540 | 8,9 | 50 |
| 240 | 1,5ч | 16,45 | 1,47 | 0,539 | 6,1 | 23 |
| 240 | 2ч | -16,45 | -1,8 | 0,539 | 5,0 | 10 |
| 240 | 2,5ч | — | — | — | — | — |
| ПАН-нить, полученная по режиму | II | |||||
| 20 | — | 16,83 | 0,8 | 0,517 | 10,2 | 144 |
| 50 | — | 16,8 | 0,77 | 0,5 | 10,1 | 152 |
| 135 | — | 16,6 | 0,77 | 0,5 | 10,1 | 152 |
| 170 | — | 16,4 | 0,7 | 0,534 | 12,7 | 174 |
| 195 | — | 16,33 | 0,6 | 0,539 | 14,8 | 200 |
| 220 | — | 16,2 | 0,575 | 0,5 | 15,2 | 223 |
| 238 | — | 16,2 | 0,515 | 0,5 | 17 | 245 |
| 240 | 0,5ч | 16,2 | 0,6 | 0,540 | 14,8 | 164 |
| 240 | 1,0ч | 16,25 | 0,9 | 0,5 | 9,8 | 60 |
| 240 | 1,5ч | -16,33 | -2 | 0, | 4,2 | 18 |
| 240 | 2,0ч | — | — | — | — | — |
ний материала ПАН в наноструктуру, промежуточную между структурами ПАН и углеродного волокна.
На дифрактограмме термостабилизированного при 240 °С волокна выявляется только интенсивный широкий максимум при значениях 263 = 26,05 градусов (рис. 3). В зависимости от режимов термостабилизации положение этого максимума на дифрактограмме несколько изменяется (табл. 2). Однако средние размеры областей когерентного рассеяния формирующейся нанодисперсной фазы («предструктуры» углеродного волокна) остаются неизменными. Разрывная прочность термостабилизированного волокна, структурные преобразования которого на стадии термостабилизации развивались отчетливо гетерогенно, уменьшается в 2—3 раза.
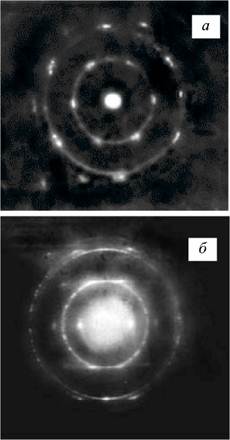
Рис. 4. Типичные микроэлектронограммы углеродного волокна, полученного при -3000 "С:
а — точечная электронограмма микрообъема волокна, характерная для крупнокристаллической структуры; б — электронограмма более дисперсной части углеродной нити
Высокотемпературную обработку волокна проводили в специальных печах косвенного нагрева, оснащенных графитовым нагревателем. Время прогрева углеродного жгута до заданной температуры в изотермическом поле печи не превышало 1 с (8). В результате высокоскоростной термомеханической обработки термостабилизированного волокна (температура 2300 °С, продолжительность изотермического нагрева -15 с) формируется структура углеродного волокна, средние размеры ОКР которой £002 ~ 2,5 нм, а межслоевое расстояние d002 ~ 0,349 нм. Причем уменьшение продолжительности термообработки в два раза практически не влияет на параметры структуры углеродного волокна.
После дополнительной высокоскоростной термообработки при -3000 °С в атмосфере аргона средние размеры областей когерентного рассеяния L002, сформировавшиеся в углеродном волокне, полученном на основе термостабилизированной ПАН-нити, процесс окисления которой развивался более активно, увеличиваются до -11 нм, а межслоевое расстояние fl?002 ~ 0,3435 нм. При исследовании тонких срезов углеродного волокна методом электронной микродифракции наряду с кольцевыми наблюдаются и точечные микро-дифрактограммы (рис. 4), характерные для графитиро-ванных материалов. Это указывает на формирование в локальных микрообъемах материала углеродной нити достаточно крупных областей когерентного рассеяния. Углеродный материал нити, полученной при -3000 °С, оказывается пространственно неоднородным по размеру областей когерентного рассеяния. Формирование крупных областей когерентного рассеяния в углеродной нити обуславливает резкое уменьшение прочности волокна.
Таким образом, в начале окислительной термообработки происходит заметное увеличение средних размеров областей когерентного рассеяния полиакри-лонитрила. Протекающие реакции циклизации, дегидрогенизации и окисления стимулируют в локальных микрообъемах ПАН-волокна фазовый переход поли-акрилонитрила в промежуточную нанодисперсную фазу структуры углеродного волокна. Режимы формования и технология получения ПАН-нити существенно влияют на закономерности перехода полиакрило-нитрила в нанодисперсную фазу, а также на средние размеры областей когерентного рассеяния и свойства углеродного волокна, получаемого в процессе высокоскоростной термообработки при -3000 °С.
1. Edie D.D. Carbon, 1998, v. 36, № 4, p. 345-362.
2. Stephen D., Frank H., Peter M.B. Polymer, 1999, № 40, p. 5531-5543.
3. Gupta A., Harrison I.R. Carbon, 1997, v. 35, № 6, p. 809— 818.
4. Fitter E., Frohs W., Heine M. Ibid., 1986, v. 24, № 4, p. 387-395.
5. Семенов П.А., Тюмещев В.А., Свиридов А.А., Подкопаев С.А., Швейкин Т.П. Ж. прикладной химии, 2003, т. 76, № 5, с. 838-841.
6. Goodhew P.J., Clarke A.J., Bailey J.A. Mater. Sci. and Eng., 1975, v. 17, № 1, p. 3-30.
7. Gupta A.K., Maiti A.K. J. Appl. Polym. Sci., 1982, v. 27, p. 2409-2414.
8. Подкопаев С.А., Шибаленков Д.Н., Тюменцев В.А., Семенов П.В., Лучко Д.В., Богословский С.А. Ж. прикладной химии, 2002, т. 75, № 2, с. 339-341.
Для подготовки данной работы были использованы материалы с сайта http://www.chem.msu.su/